Interested Article - Синдром Мартина — Белл
- 2021-10-26
- 2
Синдро́м Ма́ртина — Белл (синдром ломкой X-хромосомы, fragile X syndrome , FraX (от англ. fragile — хрупкий, ломкий)) — наследственное заболевание .
Развитие синдрома связано с экспансией единичных тринуклеотидов (ЦГГ) в Х-хромосоме и приводит к недостаточной экспрессии белка FMR1, который необходим для нормального развития нервной системы. Существует четыре основных состояния хромосомного участка, подверженного нарушениям при синдроме ломкой Х-хромосомы, которые относятся к удлинению повторяющихся последовательностей ЦГГ. Нормальное количество повторов (отсутствие синдрома) — от 29 до 31. Премутация — от 55 до 200 повторов (синдром не развивается). Полная мутация — более 200 повторов (обычно от 230 до 4000), при которой проявляется синдром. Промежуточное состояние, или аллели серой зоны, — от 40 до 60 повторов .
История
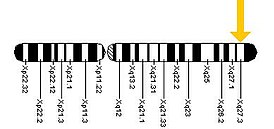
В 1943 году Джеймсом Мартином (James Purdon Martin) и впервые была описана семья, где умственная отсталость наследовалась сцепленно с полом . В этой английской семье 11 умственно отсталых детей мужского пола с психическим возрастом 2—4 года родились у интеллектуально нормальных матерей . В двух случаях относительно небольшой интеллектуальный дефицит был у женщин этого семейства . В 1969 году Hebert Lubs, проводя цитогенетическое обследование, выявил у больного синдромом Мартина-Белл вторичную перетяжку на длинном плече Х-хромосомы в локусе Xq27-28 .
Частота распространения — 1:1000—1:2000 новорожденных мальчиков.
Этиология

Синдром ломкой Х-хромосомы развивается в результате мутации гена FMR1 в Х-хромосоме. Мутация в этом гене встречается приблизительно у одного из 2000 мужчин и у одной из 259 женщин. Распространённость непосредственно заболевания — приблизительно 1 из 4000 мужчин и 6000 женщин .
Экспансия повторяющихся кодонов ЦГГ приводит к гиперметилированию ДНК в промоторе гена FMR1 и, как следствие, фактическому прекращению его экспрессии .
Как предполагают, аномальное метилирование промотора гена FMR1 в локусе Хq27.3 является причиной формирования сайта ломкости Х-хромосомы. По этому цитогенетическому признаку синдром Мартина — Белл получил своё второе название — синдром ломкой Х-хромосомы.
Мутация гена FMR1 приводит к подавлению транскрипции белка FMR1. У здоровых индивидов FMR1, как считают, регулирует значительную популяцию мРНК : FMR1 играет важную роль в обучении и запоминании, а также принимает участие в развитии аксонов , формировании синапсов , появлении и развитии нервных связей .
Наследование
Синдром ломкой Х-хромосомы — сцепленное с полом доминантное заболевание с редуцированной пенетрантностью .
Мужчины имеют одну Х-хромосому, соответственно, если она содержит мутантный аллель, у носителя развивается заболевание. Женщины несут две Х-хромосомы, таким образом, их шанс получить нормальный аллель удваивается. Женщина с мутантным геном FMR1 может иметь симптомы болезни или быть здоровой. Несмотря на то, что вторая Х-хромосома может служить резервной копией, только одна Х-хромосома активна в каждой отдельной клетке, вследствие инактивации второй .
Мужчина с ломкой Х-хромосомой не может передать её ни одному из сыновей, только всем дочерям. Женщина с одной мутантной хромосомой имеет одинаковые шансы передать её как дочерям, так и сыновьям с вероятностью 50 %. Наследование синдрома ломкой Х-хромосомы обычно увеличивается с каждым новым поколением, это явление получило название .
Патогенез
В начале 1990-х годов было осуществлено секвенирование гена синдрома Мартина — Белл. Полученные результаты показали, что в основе клинических проявлений и цитогенетически выявляемой ломкости X-хромосомы при этом заболевании лежит многократное увеличение числа тринуклеотидных повторов ЦГГ. Оказалось, что у здоровых индивидов число этих повторов в X-хромосоме колеблется от 6 до 54, а увеличение этого числа свыше 200 повторов приводит к феномену ломкой X-хромосомы и клиническому проявлению заболевания. Предмутационное состояние — когда повторов ЦГГ от 55 до 200: заболевание у таких людей в типичной форме не проявляется, но высока вероятность того, что оно проявится у их потомков.
Экспансия тринуклеотидных повторов происходит во время гаметогенеза . Переход от состояния предмутации к полной мутации возможен только при передаче гена от матери, то есть «утяжеление» аллеля происходит во время овогенеза .
Клиническая картина

Мальчики рождаются с большой массой тела — от 3,5 до 4 кг. Первым признаком, который заставляет заподозрить заболевание, является (увеличение размеров яичек) при отсутствии . Также есть определённые фенотипические признаки: большая голова с высоким и широким лбом, длинное лицо с увеличенным подбородком, несколько уплощённая средняя часть лица, тупой, слегка клювовидно загнутый кончик носа. Уши большие, иногда оттопыренные, низко расположенные. Кисти и стопы широкие, дистальные фаланги пальцев также широкие, суставы имеют повышенную подвижность. Кожа нередко гиперэластична. Часто встречаются светлоокрашенные радужные оболочки, светлые волосы. Не обязательно встречаются все признаки — могут быть один или несколько.
Неврологическая симптоматика неспецифична, определяется как и у всех детей с умственной отсталостью . Наблюдается некоторая мышечная гипотония , движений. Также могут быть глазодвигательные, пирамидные и экстрапирамидные нарушения.
Главным симптомом синдрома является интеллектуальное недоразвитие и своеобразная речь. Такие больные говорят быстро, сбивчиво, имеются выраженные эхолалии и персеверации (бормочущая речь). Степень умственной отсталости при синдроме Мартина — Белл колеблется между средней и лёгкой умственной отсталостью . Также могут быть нарушения поведения в виде агрессивности, двигательной расторможенности. В качестве одной из частых психопатологических особенностей отмечена симптоматика, напоминающая аутистическую : стереотипии , эхолалия , мутизм , самоповреждения , трудно устанавливаемый зрительный контакт и непереносимость прикосновений . Однако в отличие от аутистов, эти дети стремятся к общению . Встречаются также подпрыгивания, похлопывания руками, повороты вокруг своей оси, встряхивание кистями, «манежный» бег, разнообразные гримасы, монотонное хныканье.
Кроме вышеописанного у таких детей могут быть признаки раннего детского аутизма .
Диагностика

Синдром хрупкой Х-хромосомы диагностируется путём определения количества ЦГГ-повторов и их статуса метилирования с помощью эндонуклеазной рестрикции и саузерн-блоттинга .
Это заболевание относится к болезням экспансии (экспансия — резкое увеличение числа копий повторяющихся участков молекулы ДНК (повторы) у индивидов в последующих поколениях родословной). Феномен экспансии числа тринуклеотидных повторов (ЦГГ) был впервые обнаружен как раз при молекулярно-генетическом исследовании этого синдрома.
Ранее диагноз синдрома Мартина-Белл основывался на данных клинико-генеалогического анализа и результатах цитогенетического исследования клеток больного, выращенных на специальной среде с дефицитом фолиевой кислоты. В случае обнаружения поломок X-хромосомы в локусе Xq27.3 диагноз синдрома не вызывает сомнений.
Лечение
Лечения для синдрома ломкой Х-хромосомы не существует, однако есть надежда, что дальнейшие исследования причин заболевания предоставят новые возможности терапии. В настоящее время симптомы можно облегчить с помощью когнитивно-поведенческой терапии, специфического обучения , медикаментов и, при необходимости, лечения физических аномалий. Лица, имеющие случаи синдрома ломкой Х-хромосомы в семье, должны получить генетическое консультирование при планировании беременности .
Поскольку в эксперименте обнаружение ломкости удалось обнаружить в среде, бедной фолатами , было предложено лечить таких детей фолиевой кислотой .
Эффект от лечения у детей выражен больше, чем у взрослых: пропадает агрессия, повышается внимание, улучшается моторика и речь .
Также пробуют лечить таких больных психостимуляторами .
Примечания
- (англ.) — 2016.
- Sherman, S. Epidemiology // Fragile X Syndrome, Diagnosis Treatment and Research (англ.) / Hagerman, R. J.; Hagerman, P. J.. — 3rd. — Baltimore: Johns Hopkins University Press , 2002. — ISBN 0801868432 .
- ↑ J. Purdon Martin, Julia Bell. A pedigree of mental defect showing sex-linkage (англ.) // (англ.) (: journal. — 1943. — Vol. 6 , no. 3—4 . — P. 154—157 . — doi : . — . — PMC .
- Nolin S.L., Brown W.T., Glicksman A., et al. (англ.) // (англ.) (: journal. — 2003. — Vol. 72 , no. 2 . — P. 454—464 . — doi : . — . — PMC .
- Bassell G.J., Warren S.T. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function (англ.) // (англ.) (: journal. — Cell Press , 2008. — Vol. 60 , no. 2 . — P. 201—214 . — doi : . — .
- Garber K.B., Visootsak J., Warren S.T. (англ.) // (англ.) (: journal. — 2008. — Vol. 16 , no. 6 . — P. 666 . — doi : . — . 4 ноября 2012 года.
- ↑ Н. Н. Иванец, Ю. Г. Тюльпин, В. В. Чирко, М. А. Кинкулькина. . — М. : ГЭОТАР-Медиа, 2006. — С. . — 832 с. — ISBN 5-9704-0197-8 .
- Hagerman R.J., Berry-Kravis E., Kaufmann WE et al. Advances in the treatment of fragile X syndrome (англ.) // (англ.) (. — (англ.) (, 2009. — Vol. 123 , no. 1 . — P. 378—390 . — doi : . — . — PMC .
Ссылки
- Medportal.ru
- Пер. с англ. Н. Д. Фирсовой (2018)
- 2021-10-26
- 2























