Interested Article - Пространственное выравнивание
- 2021-03-28
- 1

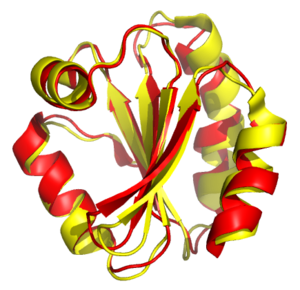
Простра́нственное выра́внивание — способ установления гомологии между двумя или более полимерными структурами на основании их трёхмерной структуры. Этот процесс обычно применяется к третичной структуре белков , но может также использоваться и для больших молекул РНК . В противоположность простому наложению структур, когда известно по крайней мере несколько эквивалентных аминокислотных остатков , пространственное выравнивание не требует никаких предварительных данных, кроме координат атомов .
Пространственное выравнивание подходит для сравнения белков с непохожими последовательностями, когда эволюционные отношения не могут быть установлены стандартными методами выравнивания последовательностей , но в этом случае необходимо принимать во внимание влияние конвергентной эволюции .
Пространственное выравнивание позволяет сравнивать две и более молекулы , для которых известны трехмерные структуры. Два основных метода их получения — рентгеноструктурный анализ и ЯМР-спектроскопия . Для пространственного выравнивания можно также использовать структуры, полученные методами предсказания структуры белка . Пространственные выравнивания особенно важны для анализа данных, полученных методами структурной геномики и протеомики, они также могут использоваться для оценки выравниваний, полученных путём сравнения последовательностей .
Данные, получаемые путём структурного выравнивания
Результатом работы программ структурного выравнивания, как правило, является совмещение наборов координат атомов и наименьшее среднеквадратическое отклонение (RMSD) между структурами. Кроме того, могут быть рассчитаны и более сложные параметры, оценивающие структурное сходство, например, . RMSD указывает на степень расхождения выравниваемых структур. Структурное выравнивание может быть затруднено из-за наличия нескольких доменов в структуре выравниваемых белков, так как изменения в относительном расположении этих доменов между двумя структурами могут искусственно изменять значение RMSD. Из структурного выравнивания непосредственно вытекает соответствующее одномерное выравнивание последовательностей, кроме того, на его основании можно рассчитать долю аминокислотных остатков, идентичных между двумя белками.
Типы сравнений
Для создания структурного выравнивания и подсчёта соответствующих значений RMSD могут быть использованы как все атомы, входящие в молекулу белка, так и их подмножества. Например, атомы боковых радикалов аминокислотных остатков учитываются не всегда, и для выравнивания могут использоваться только атомы, входящие в пептидный остов молекулы. Такой вариант выбирают, если у выравниваемых структур очень разная аминокислотная последовательность и боковые радикалы различаются у большого числа остатков. По этой причине по умолчанию методы пространственного выравнивания используют только атомы остова, вовлечённые в пептидную связь . Для большего упрощения и увеличения эффективности часто используется положение только альфа-атомов углерода , так как их положение достаточно точно определяет положение атомов полипептидного остова. Только при выравнивании очень похожих или даже идентичных структур важно учитывать позиции атомов боковых цепей. В этом случае RMSD отражает не только схожесть конформации белкового остова, но и ротамерные состояния боковых цепей. Другие способы, позволяющие снизить шум и увеличить число правильных сопоставлений, используют разметку элементов вторичной структуры , карты или паттерны взаимодействия остатков, меры степени упаковки боковых цепей и меры сохранения водородных связей .
Самый простой способ сравнить две структуры не требует выравнивания самих структур, а использует выравнивание последовательностей. Оно определяет, какие пары аминокислотных остатков сопоставлены друг другу, и затем только они используются для подсчёта RMSD. Наложение структур обычно используется для сравнения нескольких конформаций одного белка (в этом случае даже не нужно выравнивать последовательности) и для оценки качества выравниваний последовательностей, если для них известны структуры. Традиционно при наложении структур используется простой метод наименьших квадратов , в котором оптимальные повороты и трансляции находят через минимизацию суммы квадратов расстояний между всеми структурами в наложении . В недавнее время подобный поиск стал более точным благодаря методам максимального правдоподобия и байесовским методам .
Алгоритмы, основанные на многомерных поворотах и модифицированных кватернионах , были разработаны для определения топологических отношений между структурами белков без построения выравнивания последовательностей. Такие алгоритмы успешно определили канонические укладки, такие как . Метод позволяет учитывать относительные вращения доменов и другие сложные моменты структурного выравнивания .
Представление структур
Для того чтобы сравнивать структуры белков, нужно представить их в пространстве, которое не зависит от координат. Это обычно достигается с помощью матрицы «последовательность против последовательности» или серии матриц, которые включают меры сравнения, относящиеся к фиксированному пространству координат, а не абсолютные расстояния. Очевидным способом подобного представления является матрица расстояний , которая представляет собой двумерную матрицу , содержащую все попарные расстояния между некоторым набором атомов в каждой структуре (например, ). Размерность такой матрицы с увеличением числа одновременно сравниваемых структур растёт. Представив белок в виде крупных частей, таких как элементы вторичной структуры (SSEs) или другие структурные фрагменты, тоже можно получить разумное выравнивание, несмотря на потерю информации от неучтенных расстояний, так как не будет учитываться и шум от них. Таким образом, выбор способа представления белка для облегчения вычислений является критическим для разработки эффективного алгоритма выравнивания .
Вычислительная сложность
Оптимальное решение
Было показано, что оптимальное « » белковой последовательности через известную структуру и построение оптимального множественного выравнивания последовательностей являются NP-полными задачами . Однако обычная задача структурного выравнивания не является NP-полной. Строго говоря, оптимальное решение задачи структурного выравнивания белков известно только для некоторых мер сходства белковых структур — например, мер, используемых в задачах предсказания структуры белка GDT_TS и MaxSub . Такие меры могут быть оптимизированы, используя алгоритм, способный максимизировать число атомов в двух белках, которые могут быть совмещены, так как удовлетворяют предустановленному порогу на расстояние между ними. К сожалению, алгоритм оптимального выравнивания непрактичен, так как время его работы зависит не только от длин последовательностей, но и от геометрии выравниваемых белков .
Приближенное решение
Были разработаны и приближённые алгоритмы структурного выравнивания, работающие полиномиальное время и выдающие целое семейство «оптимальных» решений в пределах параметра приближения для заданной . Хотя теоретически задача приближённого структурного выравнивания белков легко даётся таким алгоритмам, они всё равно являются вычислительно затратными для масштабного анализа белковых структур. Как следствие, не существует практичных алгоритмов, которые с заданной функцией счёта сходились бы к глобальному решению выравнивания. По этой причине большинство алгоритмов являются эвристическими , но всё же были разработаны практичные алгоритмы, которые гарантируют схождение хотя бы к локальной максимизации функции счёта .
Методы
Структурное выравнивание используется как при сравнении отдельных структур или их наборов, так и при создании баз данных сравнений «все-против-всех» («all-to-all»), которые отражают различия между каждой парой структур, присутствующих в Protein Data Bank (PDB). Такие базы данных обычно используются для классификации белков по их укладке.
DALI
Одним из популярных методов структурного выравнивания является DALI ( англ. distance alignment matrix method — метод с использованием матрицы дистанционных выравниваний). В нём исходные структуры белков разбиваются на гексапептиды и через оценку паттернов контактов между фрагментами рассчитывается матрица расстояний. Элементы вторичной структуры, остатки которых являются соседними в последовательности, оказываются на главной диагонали матрицы; остальные диагонали матрицы отражают пространственные контакты между остатками, которые в последовательности не находятся рядом друг с другом. Если эти диагонали параллельны главной диагонали, то элементы вторичной структуры, которые они представляют, тоже параллельны; если они, напротив, перпендикулярны ей, то их элементы вторичной структуры антипараллельны. Такое представление интенсивно работает с памятью, так как используемая матрица симметрична относительно главной диагонали (и потому избыточна) .
Когда матрицы расстояний двух белков имеют одинаковые или похожие элементы примерно на одинаковых позициях, можно сказать, что белки имеют схожую укладку и их элементы вторичной структуры соединены петлями примерно одинаковой длины. Непосредственный процесс выравнивания DALI заключается в поиске схожестей матриц, построенных для двух белков; обычно это осуществляется с помощью серии перекрывающихся подматриц размера 6 × 6. Соответствия подматриц потом пересобираются в конечное выравнивание с помощью стандартного алгоритма максимизации счёта. Оригинальная версия DALI использует симуляцию Монте-Карло для максимизации значения пространственной схожести, являющегося функцией от расстояний между предполагаемыми соответствующими атомами. В частности, вес более отдалённых атомов внутри соответствующих элементов структуры экспоненциально занижается, чтобы уменьшить шум, вызванный подвижностью петель, искривлением спиралей и другими мелкими вариациями структур . Поскольку DALI основан на матрице расстояний типа «все-против-всех», метод может учитывать расположение элементов структур в различном порядке в двух сравниваемых последовательностях.
Метод DALI был использован для создания базы данных ( англ. Families of Structurally Similar Proteins ), в которой все известные структуры белков были попарно выровнены для определения их пространственного родства и классификации укладок .
является скачиваемой программой, использующей алгоритм DALI .
Комбинаторное расширение
Метод комбинаторного расширения ( англ. Combinational extension (СЕ) ) похож на DALI тем, что тоже разбивает каждую структуру на ряд фрагментов, которые затем пытается заново собрать в полное выравнивание. Серия попарных сочетаний фрагментов, называемых AFP ( англ. aligned fragment pairs — пары выровненных фрагментов), используется для задания матрицы сходства, через которую прокладывается оптимальный путь для определения конечного выравнивания. Только те AFP, которые удовлетворяют заданным критериям локального сходства, включаются в матрицу, что сокращает необходимое пространство поиска и увеличивает эффективность . Возможны разные меры сходства; в первоначальном виде метод CE использовал только структурные совмещения и расстояния между остатками, но со временем был расширен для использования локальных свойств, таких как вторичная структура, доступность растворителя, паттерны водородных связей и двугранные углы .
Путь, соответствующий выравниванию, рассчитывается как оптимальный путь через матрицу сходства с помощью линейного прохода через последовательности, расширяя выравнивание следующей возможной AFP с высоким счётом. Начальная AFP, инициирующая выравнивание, может быть выбрана в любой точке матрицы последовательностей. Далее происходит расширение на AFP, которая удовлетворяет заданному критерию на расстояние, ограничивающему размер гэпов (разрывов) в выравнивании. Размер каждой AFP и наибольшая длина гэпа являются необходимыми входными параметрами, но обычно устанавливаются равными эмпирически определённым значениям 8 и 30 соответственно . Подобно DALI или SSAP, CE использовался для создания базы данных классификации укладок на основе известных пространственных структур белков из PDB. Недавно PDB выпустил обновлённую версию CE, которая может определять циклические перестановки в структуре белков .
SSAP
Метод SSAP ( англ. Sequential Structure Alignment Program ) использует двойное динамическое программирование для построения структурного выравнивания, основанного на векторах «от атома к атому» в пространстве структур. Вместо альфа-атомов углерода, обычно используемых в структурных выравниваниях, SSAP задаёт свои вектора из бета-атомов для всех аминокислотных остатков за исключением глицина . Таким образом, этот метод учитывает положение ротамера каждого остатка, также как и их положение в остове. Сначала SSAP для каждого белка строит серию векторов расстояний между каждым остатком и его ближайшим, но не смежным в последовательности соседом. После этого конструируется ряд матриц, содержащих разницу векторов между соседями для каждой пары остатков, для которых строились вектора. Для каждой получившейся матрицы с помощью динамического программирования определяется ряд оптимальных локальных выравниваний. Затем полученные выравнивания складываются в обобщённую матрицу, к которой снова применяется динамическое программирование для определения полного структурного выравнивания. Изначально SSAP создавал только попарные выравнивания, но в дальнейшем был расширен и для создания множественных выравниваний . Он был применён для выравнивания типа «все-против-всех» для создания иерархической системы классификации укладок, известной как CATH, которая используется в базе данных .
Недавние разработки
Усовершенствование методов пространственного выравнивания остаётся активно исследуемой областью. Новые или модифицированные методы часто имеют преимущества над более старыми и широко распространенными техниками. Один из недавних примеров — программа ТМ-align , использующая новый метод для взвешивания матрицы расстояний, к которой потом применяется динамическое программирование . Взвешивание ускоряет сходимость динамического программирования и корректирует эффект от длины выравнивания. Тесты показали, что ТМ-align работает с более высокой точностью и скоростью, чем DALI и CE .
Однако с новыми алгоритмическими продвижениями и развитием вычислительных мощностей стало ясно, что нет универсального критерия для оптимального выравнивания. Поэтому недавние разработки сфокусированы на оптимизации конкретных параметров, таких как скорость, вычисление счетов, корреляция с альтернативными золотыми стандартами или устойчивость к погрешностям структурных данных или ab initio структурных моделей. Альтернативная методология, набирающая популярность — использование консенсуса множества методов, чтобы уточнить структурные сходства белков .
Гибкое выравнивание
Стандартные алгоритмы структурного выравнивания подразумевают жёсткость выравниваемых структур, что не отражает биологической реальности. Поэтому были разработаны алгоритмы гибкого выравнивания, которые рассматривают возможность движения двух фрагментов внутри белка относительно друг друга, а также внутренних перестановок фрагментов. Один из таких алгоритмов — FATCAT . Он использует AFP, как и CE (см. ), и пытается составить из них длинную цепочку, но при этом соединение между соседними AFP считается гибким и алгоритм изгибает его, если это улучшает наложение структур. FATCAT суммирует гэпы, повороты и простые присоединения новых пар к выровненной части в единую функцию счёта и строит выравнивание одновременно с определением участков петель, используя динамическое программирование.
Было показано, что гибкое выравнивание превосходит жёсткое выравнивание с точки зрения геометрического наложения и поиска сходств в структурах .
Непоследовательное выравнивание
Иногда белки могут содержать сходные фрагменты, расположенные в разном порядке, что не учитывается классическими алгоритмами. Методы непоследовательного выравнивания, независимые от порядка расположения элементов структуры, могут обрабатывать такие случаи. Примерами могут служить программы FATCAT, MASS , MultiProt .
Выравнивание молекулярных комплексов
В некоторых случаях есть необходимость сравнить структуры не одиночных молекул белка, а комплексов белков с белками или нуклеиновыми кислотами . Построение таких выравниваний затруднено по нескольким причинам. Во-первых, часто выровненные участки разбросаны по всему комплексу, тогда как конкретные цепи выровнены лишь частично. Во-вторых, необходимо учитывать подвижность белковых цепей, перемещения доменов и перестановки субъединиц. В-третьих, в комплексах встречаются повторы и симметрии, которые не могут быть наложены одновременно. Кроме того, большое количество выравниваемых атомов накладывает дополнительные требования на скорость вычислений. Для выполнения такой задачи алгоритм TopMatch строит точные локальные выравнивания, из которых затем конструируется полное выравнивание. Качество выравнивания оценивается по его длине и по пространственному отклонению выравниваемых структур. Воспользоваться методом можно на веб-сервисе TopMatch.
Выравнивание РНК
Крупные молекулы РНК , как и белковые молекулы, характеризуются сложной пространственной структурой, которая удерживается за счёт спаривания оснований посредством водородных связей и стэкинга . Однако для некодирующих РНК со сходными функциями получить данные геномики очень сложно, поскольку у таких молекул, как и у белков, структура последовательностей гораздо консервативнее, но при этом алфавит РНК существенно меньше (4 нуклеотида вместо 20 аминокислот), поэтому собственная информация любого нуклеотида в любой позиции ниже, чем у аминокислотного остатка .
Впрочем, в связи с возрастающим интересом к РНК и увеличением количества экспериментально установленных 3D структур РНК были разработаны методы оценки структурного сходства РНК. Один из таких методов — — разбивает каждую РНК-структуру на меньшие фрагменты, называемые общими элементами вторичной структуры (GSSU). GSSU далее подвергаются пространственному выравниванию, и эти частичные выравнивания объединяются в общее выравнивание .
— это метод для построения попарных выравниваний молекул РНК с низкой схожестью последовательностей . Этот метод отличается от методов для пространственного выравнивания белков тем, что он сам предсказывает пространственные структуры последовательностей РНК, подаваемых на вход, а не использует подаваемые на вход экспериментально установленные структуры. В то время как задача предсказания фолдинга белка в настоящее время не решена, пространственную структуру молекулы РНК без псевдоузлов предсказать можно .
Примечания
- Zhang Y. , Skolnick J. (англ.) // Proceedings of the National Academy of Sciences of the United States of America. — 2005. — Vol. 102, no. 4 . — P. 1029—1034. — doi : . — .
- ↑ Zemla A. (англ.) // Nucleic acids research. — 2003. — Vol. 31, no. 13 . — P. 3370—3374. — .
- Godzik A. (англ.) // Protein science : a publication of the Protein Society. — 1996. — Vol. 5, no. 7 . — P. 1325—1338. — doi : . — .
- Martin ACR. Rapid Comparison of Protein Structures (англ.) // : journal. — International Union of Crystallography , 1982. — Vol. 38 , no. 6 . — P. 871—873 . — doi : .
- Theobald D. L. , Wuttke D. S. (англ.) // Proceedings of the National Academy of Sciences of the United States of America. — 2006. — Vol. 103, no. 49 . — P. 18521—18527. — doi : . — .
- Theobald D. L. , Wuttke D. S. (англ.) // Bioinformatics. — 2006. — Vol. 22, no. 17 . — P. 2171—2172. — doi : . — .
- Diederichs K. (англ.) // Proteins. — 1995. — Vol. 23, no. 2 . — P. 187—195. — doi : . — .
- Maiti R. , Van Domselaar G. H. , Zhang H. , Wishart D. S. (англ.) // Nucleic acids research. — 2004. — Vol. 32. — P. 590—594. — doi : . — .
- ↑ Mount DM. Bioinformatics: Sequence and Genome Analysis. — 2nd ed. — NY, 2004. — ISBN 0879697121 .
- Lathrop R. H. (англ.) // Protein engineering. — 1994. — Vol. 7, no. 9 . — P. 1059—1068. — .
- Wang L. , Jiang T. (англ.) // Journal of computational biology : a journal of computational molecular cell biology. — 1994. — Vol. 1, no. 4 . — P. 337—348. — doi : . — .
- Siew N. , Elofsson A. , Rychlewski L. , Fischer D. (англ.) // Bioinformatics. — 2000. — Vol. 16, no. 9 . — P. 776—785. — .
- ↑ Poleksic A. (англ.) // Bioinformatics. — 2009. — Vol. 25, no. 21 . — P. 2751—2756. — doi : . — .
- Kolodny R. , Linial N. (англ.) // Proceedings of the National Academy of Sciences of the United States of America. — 2004. — Vol. 101, no. 33 . — P. 12201—12206. — doi : . — .
- Martínez L. , Andreani R. , Martínez J. M. (англ.) // BMC bioinformatics. — 2007. — Vol. 8. — P. 306. — doi : . — .
- Holm L. , Sander C. (англ.) // Science (New York, N.Y.). — 1996. — Vol. 273, no. 5275 . — P. 595—603. — .
- Holm L. , Sander C. (англ.) // Nucleic acids research. — 1997. — Vol. 25, no. 1 . — P. 231—234. — .
- Holm L. , Park J. (англ.) // Bioinformatics. — 2000. — Vol. 16, no. 6 . — P. 566—567. — .
- ↑ Shindyalov I. N. , Bourne P. E. (англ.) // Protein engineering. — 1998. — Vol. 11, no. 9 . — P. 739—747. — .
- Prlic A. , Bliven S. , Rose P. W. , Bluhm W. F. , Bizon C. , Godzik A. , Bourne P. E. (англ.) // Bioinformatics. — 2010. — Vol. 26, no. 23 . — P. 2983—2985. — doi : . — .
- Taylor W. R. , Flores T. P. , Orengo C. A. (англ.) // Protein science : a publication of the Protein Society. — 1994. — Vol. 3, no. 10 . — P. 1858—1870. — doi : . — .
- Orengo C. A. , Michie A. D. , Jones S. , Jones D. T. , Swindells M. B. , Thornton J. M. (англ.) // Structure (London, England : 1993). — 1997. — Vol. 5, no. 8 . — P. 1093—1108. — .
- Zhang Y. , Skolnick J. (англ.) // Nucleic acids research. — 2005. — Vol. 33, no. 7 . — P. 2302—2309. — doi : . — .
- Zhang Y. , Skolnick J. (англ.) // Proteins. — 2004. — Vol. 57, no. 4 . — P. 702—710. — doi : . — .
- Barthel D. , Hirst J. D. , Błazewicz J. , Burke E. K. , Krasnogor N. (англ.) // BMC bioinformatics. — 2007. — Vol. 8. — P. 416. — doi : . — .
- Ye Y. , Godzik A. (англ.) // Nucleic acids research. — 2004. — Vol. 32. — P. 582—585. — doi : . — .
- J. Xiang, M. Hu. // 2008 2nd International Conference on Bioinformatics and Biomedical Engineering. — 2008-05-01. — P. 21–24. — doi : .
- Dror O. , Benyamini H. , Nussinov R. , Wolfson H. (англ.) // Bioinformatics. — 2003. — Vol. 19 Suppl 1. — P. 95—104. — .
- Shatsky M. , Nussinov R. , Wolfson H. J. (англ.) // Proteins. — 2004. — Vol. 56, no. 1 . — P. 143—156. — doi : . — .
- Sippl M. J. , Wiederstein M. (англ.) // Structure (London, England : 1993). — 2012. — Vol. 20, no. 4 . — P. 718—728. — doi : . — .
- Torarinsson E. , Sawera M. , Havgaard J. H. , Fredholm M. , Gorodkin J. (англ.) // Genome research. — 2006. — Vol. 16, no. 7 . — P. 885—889. — doi : . — .
- Hoksza D. , Svozil D. (англ.) // Bioinformatics. — 2012. — Vol. 28, no. 14 . — P. 1858—1864. — doi : . — .
- Cech P. , Svozil D. , Hoksza D. (англ.) // Nucleic acids research. — 2012. — Vol. 40. — P. 42—48. — doi : . — .
- Havgaard J. H. , Lyngsø R. B. , Stormo G. D. , Gorodkin J. (англ.) // Bioinformatics. — 2005. — Vol. 21, no. 9 . — P. 1815—1824. — doi : . — .
- Mathews D. H. , Turner D. H. (англ.) // Current opinion in structural biology. — 2006. — Vol. 16, no. 3 . — P. 270—278. — doi : . — .
- 2021-03-28
- 1