Novo Nordisk
- 1 year ago
- 0
- 0

Сборка транскриптома de novo — метод сборки последовательностей транскриптома , который осуществляется без картирования на референсный геном. Из коротких фрагментов (ридов или прочтений), полученных при секвенировании , воссоздаются отдельные последовательности РНК или транскрипты.
В период с 2008 по 2018 год в связи с разработкой новых технологий произошло значительное снижение стоимости секвенирования. До технологического прорыва секвенирование транскриптомов производилось только для тех организмов, которые представляли большой интерес для научных исследований. Однако с развитием технологий секвенирования следующего поколения и методов RNA-seq стало возможно секвенировать транскриптомы сложных, полиплоидных немодельных организмов. Создание сборок de novo для модельных организмов полезно для обнаружения новых изоформ существующих аннотированных генов, альтернативных событий сплайсинга и новых транскрибируемых генов в ответ на специфическое воздействие.
Геномы большинства организмов сложны для сборки из-за большого размера, а также повсеместно встречающихся повторов . Транскриптомы же организмов, состоящие из последовательностей РНК, из которых в процессе сплайсинга вырезаются интроны, гораздо меньше по размеру и количеству повторов. Поэтому гораздо дешевле секвенировать транскриптом, чем геном. Последовательность генома — эта наиболее полная информация об организме, однако реализация генетической программы порой гораздо важнее и инетереснее, чем сама программа. Поэтому узнать о том, какие участки генома транскрибируются и какие продукты они дают, можно при помощи технологии секвенирования RNA-seq. Транскриптомные данные почти всегда необходимы для точной аннотации генома. Что касается технической стороны обработки данных, алгоритмы сборки геномов плохо работают в случае неравномерного покрытия ридами, а в случае транскриптома гены могут иметь очень разное покрытие из-за разницы в уровнях эксрессии.
De novo сборка транскриптома не требует наличия референсного генома. Учитывая, что большинство организмов на данный момент еще не отсеквенированы, de novo сборка транскриптома таких организмов может быть использована как первый этап в их изучении. Например, для изучения и сравнения транскриптомов между организмами, а также для анализа дифференциальной экспрессии при различных воздействиях на организм. Иногда бывает полезно собирать транскриптом de novo даже при наличии референсного генома, так как при этом можно обнаружить транскрибируемые участки, последовательности которых отсутствуют в геномной сборке. Имея de novo сборку и референсный геном можно детектировать транскрипты экзогенного происхождения. Самое важное отличие de novo сборки в том, что при этом не требуется выравнивать последовательности и решать проблемы поиска или предсказания сайтов сплайсинга, помимо этого удается собирать транскрипты, полученные в результате транссплайсинга . Однако сборка de novo является алгоритмически сложным и вычислительно затратным процессом. Также данный подход отличается высокой чувствительностью к ошибкам.
Для секвенирования транскриптома используются стандартные протоколы подготовки образцов, в ходе которых выделяют все возможные последовательности РНК (тотальная РНК), уменьшают количество рибосомной РНК , производят обратную транскрипцию , фрагментируют кДНК , лигируют адаптеры, амплифицируют и секвенируют короткие фрагменты кДНК.
В случае de novo сборки транскриптома важно принимать во внимание некоторые особенности пробоподготовки. Во-первых, в одном организме транскриптом клеток может значительно различаться в зависимости от ткани, времени взятия образца, а также наличия того или иного стресса. Для сборки транскриптома de novo неизвестного организма необходимо иметь информацию о всех экспрессирующихся генах. Для этого необходимо секвенировать как можно больше органов и тканей в условиях различных стрессов. Во-вторых, так как гены различаются по уровню экспрессии, то в составе тотальной РНК одних транскриптов будет больше, а других меньше. Соответственно больше всего ридов будет получено от высокоэкспрессирующихся генов, в то время как низкоэкспрессирующиеся гены могут быть неотсеквенированы. Из этого вытекает еще одна особенность сборки транскриптома de novo : глубина секвенирования должна быть довольно высока, чтобы детектировать даже низкоэкспрессирующиеся гены. Существует экспериментальная процедура нормализации библиотеки кДНК, которая позволяет увеличить среднее покрытие. Этот метод основан на действии дуплекс-специфической нуклеазы. При этом молекулы кДНК, полученные в ходе обратной транскрипции, нагревают, чтобы они денатурировали. При ренатурации одноцепочечные молекулы кДНК высокоэкспрессированных транскриптов быстрее образуют дуплексы, которые затем расщепляются специальной нуклеазой. Так уменьшается количество кДНК высокоэкспрессируемых генов и увеличивается вероятность, что при данной глубине секвенирования получится увеличить покрытие низкоэкспрессируемых генов.
В процессе секвенирования могут возникать ошибки при прочтении.
Качество секвенирования оценивается с помощью метрики (Q score). Она рассчитывается для каждого нуклеотида по формуле , где P — это вероятность ошибки. При Q > 20 можно говорить о хорошем качестве прочтения нуклеотида, то есть вероятность того, что он неверно отсеквенирован составляет 1 %. Первичный анализ данных секвенирования (QC report) проводится в программе FastQC. Далее по порогу Q score можно отсекать нуклеотиды с низким качеством с концов прочтения (там качество заметно ниже), а также удалять последовательности адаптеров. Эта процедура называется триммированием.
Проблема перепредставленности некоторых транскриптов и, соответственно, проблема неравномерности покрытия решается не только с помощью экспериментальной процедуры, но и методом цифровой нормализации (digital normalization) . При этом создается хеш-таблица : последовательность k-мера и его представленность (количество во всех ридах). Далее на основании этой таблицы для каждого рида рассчитывается медианное значение покрытия по k-мерам, из которых он состоит. Устанавливается порог по покрытию, по которому прочтения с медианным значением выше данного отбрасываются. Таким образом удаляются риды с очень большим покрытием.
Алгоритмы сборки транскриптома de novo очень схожи с таковыми для сборки генома. Их можно разделить на две группы:
В отличие от сборки генома, при сборке транскриптома возникает задача восстановить множество нуклеотидных последовательностей, имеющих различную длину, а не одну исходную последовательность.
Алгоритмы OLC работают непосредственно с ридами и производят с ними следующие операции :
OLC подходы были разработаны для сборки длинных прочтений, созданных по методике Сэнгера, и были широко распространены до появления секвенирования следующего поколения. Сейчас однако есть популярные пакеты, использующие данные алгоритмы для сборки транскриптомных и геномных последовательностей.
С развитием технологий секвенирования следующего поколения, получение фрагментов (ридов) стало на порядок дешевле, но размер фрагментов стал меньше. Для сборки из коротких прочтений было предложено использовать графы де Брёйна. Вершинами графа де Брёйна являются возможные k -меры (строки длины k ), выделенные из исходных прочтений. Два k -мера, соединяются в графе ребром, если они являются префиксом и суффиксом k+1 -мера, также представленного в исходных прочтениях. Ребром является k+1 -мер.
Оптимальное значение k для сборки зависит от длины прочтения, глубины секвенирования, частоты ошибок и сложности транскриптома конкретного вида. Для низкоэкспрессируемых генов с маленьким покрытием, для которых риды слабо перекрываются для улучшения качества сборки подходит уменьшенное значение k . В то время как большие значения k позволяют разрешать повторы и участки с ошибками. Оптимальные значения k лежат в пределе от 21 до 50.
Далее производятся четыре типа упрощений графа: сжатие путей, удаление ошибок, раздвоение вершин, из которых выходит несколько ребер и, если доступны парные чтения, разрешение небольших повторов. После этого риды накладываются на граф де Брёйна и происходит восстановление последовательностей контигов: при этом последовательно обходят все ребра графа.
После построения графа требуется провести дополнительные процедуры по разрешению повторов. Повтор в транскриптоме изменяет граф де Брёйна, склеивая участки графа, соответствующие различным транскриптам . Разрешение повторов — это этап, на котором происходит попытка определить, какой путь в графе на самом деле содержит повтор. Затем происходит разделение повтора, при котором создаётся по копии повтора для каждого истинного пути.
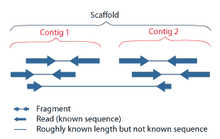
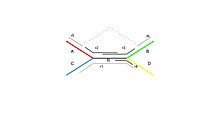
Рассмотрим пример повтора в графе де Брёйна, представленном на Рисунке 1. Сборщик использует информацию об имеющихся ридах, чтобы оценить возможность существования каждого из четырёх вариантов. Длинный рид r1 позволяет однозначно определить, что повтор имеется в последовательностях ARB и CRD, а не в ARD и CRB. Об этом же свидетельствуют риды r5 и r6. Короткие риды r2, r3, r4 не позволили бы однозначно разрешить повтор. Чем длиннее риды, тем больше повторов мы сможем разрешить. Однако при секвенировании следующего поколения получаются короткие риды. Возможность разрешить повторы появляется при использовании парных ридов.
События альтернативного сплайсинга существенно усложняют процесс сборки, причем как de novo , так и при наличии референсной последовательности. Процедура определения истинных изоформ осуществляется уже после сборки контигов. Как и в случае повторов парные чтения дают ключевую информацию о возможных изоформах. Эту информацию удобно представлять в виде сплайс-графов, в вершинах которых находятся экзоны, а ребра отображают возможные соединения между ними.
Алгортимы, основанные на перекрывании ридов, менее чувствительны к ошибкам, однако для построения графа требуется больше вычислительной мощности. На время построения графа чтений негативно влияет число, а не длина ридов. Ключевой параметр данных алгоритмов — длина перекрытия.
Алгоритмы, основанные на графах де Брейна, очень чувствительны к повторам и ошибкам в чтениях. Одна ошибка в чтении создает k ошибочных узлов. Использование этих алгоритмов позволяет экономить память (большинство k -меров встречается во многих ридах). Также упрощается работа с повторяющимися участками. Помимо этого есть возможность отсеивать ошибки уже на начальной стадии обработки данных. Ключевой параметр данных алгоритмов — k , длина k -мера.
После того, как сборка последовательностей была создана, необходимо оценить ее качество. При оценки результатов сборки используются не один параметр, а принимаются во внимание сразу несколько показателей. Программы-сборщики для оценки качества сборки производят следующий анализ :
Метрики для оценки качества можно разделить на две группы — статистические показатели и метрики, основанные на аннотации. Первая группа была разработана раньше и используется для оценки сборки генома. К ней относят процент картировавшихся прочтений (то есть использующихся при сборке), число контигов, N50, среднее покрытие контигов, число контигов > 1000 пар нуклеотидов. Однако для использования таких метрик, как N50 и число контигов необходимо иметь оценки размера исследуемого транскриптома. Метрики, основанные на аннотации, включают в себя число находок при прямой и обратной аннотации, а также еще несколько дополнительных показателей. Ortholog hit ratio (OHR) или отношение длины участка контига, картировавшегося на последовательность из референсной базы, к длине всей этой последовательности позволяет оценить полноту сборки. Данная метрика используется при прямой аннотации. Чем она больше, тем более полная сборка в итоге была создана. При обратной же рассчитывается Collapse Factor(CF), который может быть полезен при сборке полиплоидных организмов или организмов с большим числом паралогов. При сборке несколько одинаковых контигов, которые на самом деле соответствуют паралогам, они могут быть расценены как ошибка секвенирования и «сколлапсироваться» в один единственный контиг. Эту ошибку можно зафиксировать, если при обратной аннотации (при этом референсная база белков должна быть из относительно близкого организма) одному контигу соответствует несколько белков. Collapse Factor(CF) как раз и оценивает, сколько находок соответствует контигу. Чем больше эта величина, тем хуже качество сборки из-за ее пересобранности.
Следует аккуратно выбирать метрику, а также и данные, на которых эта метрика рассчитывается. Для расчета процента картирующихся на сборку ридов, N50, среднего покрытия, среднего OHR используют полный набор результатов работы сборщика (то есть контиги, а также и риды, которые в сборку не попали — синглетоны). Только контиги используют для подсчета совпадений с референсными базами при прямой и обратной аннотации, а также при подсчете Collapse Factor(CF).
SeqMan NGen, входящий в состав программного обеспечения DNASTAR’s, включает сборщик транскриптома de novo для различных по размеру наборов данных транскриптома. В основе SeqMan NGen алгоритм, который использует RefSeq для идентификации и слияния транскриптов и автоматически аннотирует собранные транскрипты с использованием собственного инструментария аннотации DNASTAR для идентификации и выделения уже известных и новых генов .
SOAPdenovo-Trans является сборщиком транскриптома de novo, созданным на основе другого сборщика — SOAPdenovo2, предназначенного для сборки транскриптома с альтернативным сплайсингом и разным уровнем экспрессии. SOAPdenovo-Trans обеспечивает более полный способ построения полноразмерных наборов транскриптов по сравнению с SOAPdenovo2 .
Velvet выделяет для каждого чтения k-меры (все возможные последовательности длины k), после чего осуществляет сборку контигов на основе построения ориентированного графа де Брёйна. Данный алгоритм хорош для очень коротких (20-50 пн) парно-концевых чтений. Для таких ридов Velvet способен создавать контиги с N50 до 50 кб, используя данные прокариот, и до 3 кб в случае искусственных бактериальных хромосом. Для данных Solexa без парных прочтений Velvet создает контиги с N50 всего 8 кб для прокариот и 2 кб для искусственных бактериальных хромосом .
Oases — пакет программ, который предназначен для эвристической сборки транскриптов в отсутствии референсного транскриптома. Использует хеш-таблицу, динамическую фильтрацию шума (удаление «островных контигов», то есть не имеющих соседних контигов в графе де Брёйна и длиной менее 150 нуклеотидов), учитывает события альтернативного сплайсинга и осуществляет эффективное слияние нескольких сборок. Может использовать парно-концевые чтения, а также длинные последовательности для построения изоформ транскриптов .
ABySS — ещё один сборщик, использующий парно-концевые чтения. Trans-ABySS — пакет программ, реализованных на Python и Perl, для сборки данных РНК-секвенирования. Может применяется к сборкам, сгенерированным в широком диапазоне k-значений. Сначала Trans-ABySS сводит набор данных к более мелким наборам контигов, находит события альтернативного сплайсинга, включая пропуски экзонов, образование новых экзонов, сохранение интронов, формирование новых интронов и альтернативное сращивание экзонов, после чего объединяет полученные сборки. Также способен оценивать уровень экспрессии генов, идентифицировать потенциальные сайты полиаденилирования и возможные химерные гены, образованные в результате слияния нескольких генов .
Этот метод сборки транскриптома делит данные РНК-секвенирования на множество независимых графов де Брёйна (один граф соответствует одному экспрессируемому гену), после чего с помощью параллельных вычислений строит транскрипты из таких графов, включая альтернативные сплайсоформы. Может использовать как парно-концевые чтения, так и одиночные. Отличается понятным интерфейсом, который почти не требует никакой настройки параметров. Trinity состоит из трех независимых программных модулей, которые используются последовательно для создания транскриптов :
Inchworm осуществляет сборку данных РНК-секвенирования в последовательности транскриптов. Часто генерирует полноразмерные транскрипты для преобладающих изоформ, а для альтернативно сплайсированных вариантов транскриптов выдает только уникальные участки .
Сhrysalis кластеризует контиги, полученные с помощью модуля Ichworm, и строит полные графы де Брёйна для каждого кластера .
Batterfly анализирует графы де Брёйна, после чего выводит все возможные последовательности транскриптов. Сначала объединяет вершины в однозначных путях в графе для получения вершин, соответствующим более длинным последовательностям, и удаляет ребра, поддерживаемые небольим числом чтений. Затем отслеживает пути ридов в полученном графе и выдает последовательности транскриптов .
Данный метод обладает двумя алгоритмами сборки контигов:
Первый алгоритм, «исключающий», собирает контиги при большом значении k. В этому случае гены с высоким уровнем экспрессии соберутся наилучшим образом. Использованные риды удаляются и сборка повторяется, но уже с меньшим значением k. Это приводит к сборке генов с меньшим уровнем экспрессии. Такая процедура проводится несколько раз. Конечная сборка формируется при объединении контигов, полученных при разных сборках .
Второй алгоритм, «смешивающий», не удаляет чтения на каждом этапе (для различных k используется весь набор чтений). В таких условиях одинаковые контиги могут образоваться несколько раз. Для устранения такой избыточности контиги выравниваются сами на себя, и короткие и изюыточные контиги удаляются .
На вход алгоритму подается фрагменты выровненных на геном последовательностей кДНК. Первый шаг в сборке фрагментов — идентификация пар несовместимых фрагментов, образованных из различных изоформ мРНК, полученных в результате сплайсинга. Неперекрывающиеся фрагменты являются совместимыми. Перекрывающиеся фрагменты принимаются за совместимые, если их перекрывания содержат полностью идентичные интроны. Строится граф перекрытий, в котором вершины соответствуют фрагменту, а ребра между двумя вершинами указывают на то, что они совместимы и имеют перекрывания. Затем граф сокращается в результате чего происходит частичное упорядочивание фрагментов. Далее алгоритм находит минимальное множество путей такое, чтобы любая вершина входила по крайней мере в один путь. По теореме Дилуорса такое множество может быть построено, если найти максимально число фрагментов, каждый из которых не совместим с остальными из этого множества, то есть построить антицепь. Достроив каждый фрагмент антицепи до пути получим искомое покрытие. После чего фрагменты с помощью метода максимального правдоподобия распределяются по найденным транскриптам .