Метод дыхания
- 1 year ago
- 0
- 0
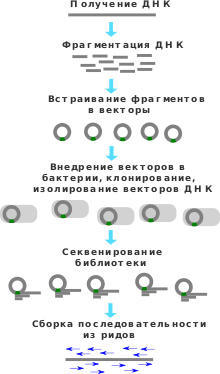

Метод дробовика ( англ. Shotgun sequencing ) — метод, используемый для секвенирования длинных участков ДНК . Суть метода состоит в получении случайной массированной выборки клонированных фрагментов ДНК данного организма, на основе которых может быть восстановлена исходная последовательность ДНК .
Предпосылкой для возникновения метода дробовика являлся тот факт, что первые методы секвенирования были способны восстанавливать лишь небольшие последовательности ДНК порядка 1000 нуклеотидов , следовательно, для секвенирования более длинных последовательностей требовалось разработать новый подход. При секвенировании методом дробовика ДНК случайным образом фрагментируется на мелкие участки, которые затем секвенируют любым доступным методом, например, методом секвенирования по Сэнгеру . Полученные перекрывающиеся случайные фрагменты ДНК затем собирают с помощью специального программного обеспечения в одну целую последовательность .
Метод дробовика использовался при получении первых полных геномов организмов .
Для примера, допустим, что имеются два случайных фрагмента, полученных методом дробовика:
| Цепь | Последовательность |
|---|---|
| Первоначальная |
AGCATGCTGCAGTCATGCTTAGGCTA
|
| Первый фрагмент |
AGCATGCTGCAGTCATGCT-------
|
| Второй фрагмент |
AGCATG--------------------
|
| Восстановленная последовательность |
AGCATGCTGCAGTCATGCTTAGGCTA
|
Данный пример является крайне упрощенным. Однако он отражает одну из важнейших особенностей процесса секвенирования генома методом дробовика. А именно, ни одно из четырёх прочтений, представленных в таблице, не покрывает полностью всю исходную последовательность. Однако исходная последовательность может быть восстановлена исходя из того, что каждый нуклеотид из исходной последовательности встречается хотя бы в одном прочтении, а за счет частичного перекрытия прочтений — более чем в одном. .
При секвенировании методом дробовика реальных молекул ДНК получаются миллионы прочтений , некоторые из которых могут содержать ошибки, которые затем должны быть собраны в исходную последовательность. Разумеется, работа такого объёма не может быть проделана вручную, поэтому для сборки последовательности ДНК из прочтений используется специальное программное обеспечение . Задача усложняется тем, что ДНК часто содержит повторяющие последовательности , а значит, похожие прочтения могут быть получены из удаленных друг от друга частей ДНК .
Для того, чтоб справиться с этой проблемой, секвенирование обычно проводят таким образом, чтобы каждый нуклеотид исходной последовательности встречался не в одном, а сразу во многих прочтениях. Так, например, при секвенировании генома человека использовалось 12-кратное покрытие, то есть каждый нуклеотид в среднем встречался в 12 прочтениях .
Идея использования метода дробовика для секвенирования малых геномов (4000—7000 т.п.н.) была предложена в 1979 году . А два года спустя — в 1981 году — метод дробовика был впервые применен на практике для секвенирования полного генома вируса мозаики цветной капусты .
Процесс секвенирования методом дробовика состоит из нескольких этапов. Сначала секвенируемая ДНК подвергается амплификации . Полученные копии ДНК разрезаются на фрагменты с помощью сайт-неспецифичных нуклеаз. Сайт-неспецифичность важна для того, чтобы получались перекрывающиеся фрагменты . Из полученных фрагментов строится геномная библиотека путём встраивания фрагментов в некоторый вектор . Из полученной геномной библиотеки случайным образом выбирается некоторое подмножество фрагментов, каждый из которых секвенируется, например, методом Сэнгера . Затем с помощью специального программного обеспечения из полученных нуклеотидных последовательностей фрагментов, называемых прочтениями, собирается нуклеотидная последовательность исходной ДНК .
В процессе сборки исходной последовательности ДНК перекрывающиеся прочтения собираются в более крупные последовательности, называемые контигами. Контиги представляют из себя непрерывные части восстанавливаемой последовательности ДНК. Контиги в свою очередь объединяются в ещё более крупные последовательности — скаффолды — которые уже не обязательно являются непрерывными частями исходной ДНК и могут содержать пропуски. Если секвенирование проводилось , то расстояние между контигами в скаффолде может быть выведено на основании информации о позиции спаренных прочтений . В зависимости от расстояния между контигами могут быть использованы различные методы для заполнения пропусков в скаффолдах. Если зазор мал (5—20 т.п.н.), то данная область амплифицируется с помощью ПЦР , а затем секвенируется. Если зазор большой (> 20 т.п.н.), то пропущенный фрагмент клонируют в специальных векторах, таких как искусственная бактериальная хромосома , с последующим секвенированием вектора .
По мере того, как стали секвенировать все более и более длинные последовательности ДНК, стало понятно, что полезно бывает секвенировать обе цепи ДНК. Во-первых, нередки случаи, когда из-за особенностей конформации ДНК определение нуклеотида на некоторой позиции на одной из цепей крайне затруднительно, тогда как на второй цепи нуклеотид в той же позиции может быть легко определён. Во-вторых, информация о взаимном расположении спаренных прочтений может быть использована для определения расстояния между контигами в скаффолде. Модификация метода дробовика, при которой секвенируются обе цепи ДНК, называется методом секвенирования парных прочтений или «двуствольным» методом дробовика. Данный метод получил широкое распространение и использовался, в частности, при секвенировании генома человека .
При секвенировании парных прочтений ДНК разрезается на случайные фрагменты, которые затем группируются по весу (обычно 2, 10, 50 и 150 т.п.н.) и клонируются в векторах . Клоны секвенируют с обоих концов с использованием метода обрыва цепи , в результате которого образуются две коротких последовательности. Каждая последовательность называется конечным прочтением или просто прочтением, а две считанные последовательности с одного и того же клона — парными концевыми. Так как длина прочтений при использовании метода обрыва цепи обычно не превышает 1000 пар оснований, то во всех, кроме самых маленьких клонов, парные концы будут перекрываться редко .
Первое опубликованное описание использования метода секвенирования парных концов датировано 1990 годом . Эта работа была посвящена секвенированию человеческого гена гипоксантин-гуанинфосфорибозилтрансферазы , но парные концы в ней использовались лишь для устранения пропусков в последовательности после применения метода дробовика в его классической форме. В 1991 году было опубликовано первое теоретическое описание секвенирования парных концов в его полноценной форме , которое предполагало использование фрагментов постоянной длины. В то время считалось, что при секвенировании парных концов оптимально использовать фрагменты, длина которых втрое больше длины прочтений. В 1995 году было показано , что при секвенировании парных концов возможно использовать фрагменты разных размеров, тем самым продемонстрировано, что данный подход может быть использован для секвенирования длинных последовательностей ДНК. Впоследствии данный подход активно использовался при секвенировании геномов различных организмов: генома гемофильной палочки в 1995 году , генома дрозофилы ( плодовой мушки ) в 2000 году и наконец генома человека в 2001 году.
Покрытие — это среднее число прочтений, покрывающих одну позицию в реконструированной последовательности. Оно может быть рассчитано исходя из длины исходного генома ( ), количества прочтений( ), и средней длины прочтения ( ), как: . Также иногда покрытием называется доля позиций генома, покрываемых прочтениями. Высокое покрытие в методе дробовика необходимо, поскольку оно позволяет избавиться от ошибок сборки, связанных с наличием в ДНК повторяющихся последовательностей .
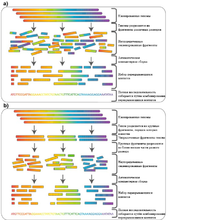
Теоретически метод дробовика может быть применен к геномам любого размера, однако изначально возможность его реального применения для полногеномного секвенирования ставилась под сомнение и из-за технических сложностей, возникающих при обработке больших объёмов данных, и из-за дополнительных сложностей, возникающих в связи с наличием огромного количества повторяющихся участков в больших геномах . Возникновение метода иерархического секвенирования сделало возможным применение метода дробовика к большим геномам на практике.
Амплифицированный геном сначала режут на крупные куски (50—200 т.п.н.) и клонируют в бактериальном хозяине с использованием искусственной бактериальной хромосомы . Поскольку несколько копий генома были разрезаны случайным образом, фрагменты, также именующиеся BAC-контигами, содержащиеся в этих клонах, имеют разные концы, а значит можно найти скаффолд, имеющий удовлетворительное и покрывающий весь геном целиком. Такой скаффолд называется покрывающим путем .

После того как покрывающий путь найден, BAC-контиги, формирующие этот путь, режутся случайным образом на более мелкие фрагменты, которые затем секвенируются с помощью метода дробовика. Хотя нуклеотидные последовательности BAC контигов неизвестны, можно определить их расположение друг относительно друга, а эту информацию впоследствии использовать для построения покрывающего пути .
Перекрывающиеся клоны могут быть идентифицированы несколькими способами. Один из способов — это использование небольшой радиоактивно или химически помеченной последовательности ДНК (STS). Такая последовательность гибридизируется на микрочипе , на котором воспроизводятся клоны . Таким образом идентифицируются все клоны, содержащие помеченную последовательность. Конец одного из этих клонов секвенируется и используется как новая STS-последовательность. Такой итеративный процесс называется хромосомной ходьбой .
Другой способ идентификации пересекающихся клонов заключается в использовании ферментов рестрикции . Определённый участок генома обрабатывается набором нуклеаз рестрикции, после чего производится сравнение размеров полученных фрагментов ДНК. Это позволяет построить рестрикционную карту, на которой указано положение каждого сайта рестрикции относительно других участков . Такой метод геномного отображения называется рестрикционным картированием, поскольку он идентифицирует набор сайтов рестрикции, содержащихся в каждом клоне .
Необходимость построения обширной BAC-библиотеки и выбора покрывающего пути делает метод иерархического секвенирования значительно более медленным и трудоёмким в сравнении с полногеномным секвенированием методом дробовика. И теперь, когда технологии позволяют производить необходимые объёмы вычислений достаточно быстро, а данные стали достаточно достоверными, полногеномное секвенирование методом дробовика вытесняет иерархическое секвенирование, так как является более эффективным как из соображений скорости, так и из соображения издержек .
Классический метод дробовика был основан на методе Сэнгера и являлся наиболее передовым методом секвенирования геномов приблизительно до 2005 года. Метод дробовика применяется и по сей день, однако на смену ему пришли новые технологии секвенирования, в отношении которых часто используется собирательное название технологии секвенирования нового поколения . Эти технологии производят более короткие прочтения (порядка 25—500 bp), но с очень высокой скоростью (порядка миллиона прочтений в день) . Как следствие, увеличивается , но процесс сборки генома из прочтений становится более вычислительно трудоёмким. Итого методы секвенирования нового поколения в сравнении с методом дробовика требуют больших вычислительных ресурсов, однако позволяют получить последовательность полного генома за более короткий срок .