Interested Article - Буллёзный эпидермолиз
- 2020-09-20
- 1
Буллёзный эпидермолиз (БЭ) — группа генетически и клинически гетерогенных заболеваний, характеризующаяся образованием пузырей и эрозий на коже и слизистых оболочках, ранимостью кожи и её чувствительностью к незначительной механической травме («механобуллезная болезнь») .
БЭ делится на 3 основных типа: простой, пограничный, дистрофический. Отдельно выделен синдром Киндлер . Разделение происходит в зависимости от уровня образования пузырей в слоях кожи на ультраструктурном уровне. При простом типе пузыри образуются в верхних слоях эпидермиса, при пограничном — на уровне светлой пластинки (lamina lucida), при дистрофическом — в верхней части сосочкового слоя дермы, ниже плотной пластинки (lamina densa). При синдроме Киндлер образование пузырей может быть на разных уровнях . Каждый основной тип БЭ подразделяется на подтипы, которых описано более 30, на основании фенотипа, характера наследования и генотипа.
Наследуется БЭ как по аутосомно-доминантному, так и по аутосомно-рецессивному типу. Частота встречаемости различных типов БЭ варьирует от 1:30000 до 1:1000000 и также зависит от популяции . Заболевание является результатом мутаций в более чем 10 генах, кодирующих белки, расположенные в различных слоях кожи.
История
Термин «буллёзный эпидермолиз» впервые был использован в 1886 году немецким дерматологом Генрихом Кёбнером , хотя случаи, схожие с данным диагнозом, описывались и до него. В 1962 году Р. В. Пирсоном была разработана первая схема классификации БЭ, основанная на применении трансмиссионной электронной микроскопии. Были выделены три основных типа: эпидермолитический, люцидолический и дермолитический, исходя из уровня образования пузырей на ультраструктурном уровне. В 1980-х годах была разработана техника иммунофлюоресцентного исследования образцов кожи, расширенная впоследствии иммуногистологическими методами (иммуногистохимией и иммунофлюоресценцией). С 1990-х годов почти для каждого подтипа БЭ были определены мутации в конкретных генах структурных белков кожи, которые изменяются при БЭ.
Классификация
Исторически сложилось так, что буллёзный эпидермолиз был классифицирован в соответствии с морфологией кожи. Наследственные варианты БЭ в настоящее время разделяются на три большие группы в соответствии с уровнем образования пузырей в тканях, отдельно выделен синдром Киндлер:
- простой буллезный эпидермолиз (ПБЭ),
- пограничный буллезный эпидермолиз (ПоБЭ),
- дистрофический буллезный эпидермолиз (ДБЭ),
- синдром Киндлер (разный уровень образования пузырей).
| Основной тип БЭ | Основные подтипы БЭ | Белки-мишени |
|---|---|---|
| Простой БЭ (ПБЭ) | Супрабазальный ПБЭ | плакофилин-1; десмоплактин; возможно другие |
| Базальный ПБЭ | кератин 5, кератин 14, α6β4-интегрин | |
| Пограничный БЭ (ПоБЭ) | ПоБЭ, подтип Херлитца | ламинин-332 (ламинин-5) |
| ПоБЭ, другие | ламинин-332; коллаген XVII типа; α6β4-интегрин | |
| Дистрофический БЭ (ДБЭ) | доминантный ДБЭ | коллаген VII типа |
| рецессивный ДБЭ | коллаген VII типа | |
| Синдром Киндлер | - | киндлин-1 |
Эпидемиология
Частота встречаемости БЭ варьирует от 1:30000 до 1:100000 человек. По данным Национального регистра БЭ (США), распространенность всех типов БЭ составляет 8,22:1 млн человек . Распространенность ПБЭ — 10,75 на 1 000 000, ПоБЭ — 2,04 на 1 000 000, ДДБЭ — 2,86 и на 1 000 000, РДБЭ — 2,04 на 1 000 000 человек.
Генетика
БЭ наследуется по аутосомно-доминантному и аутосомно-рецессивному типу. При БЭ мутации обнаруживаются в более чем 10 генах. Описаны различные виды мутаций — мисенс-мутации, нонсенс-мутации, делеции, мутации рамки считывания, инсерции, мутации сайта сплайсинга.
При наиболее распространенных подтипах ПБЭ обнаруживаются мутации в генах KRT5 и KRT14, примерно в 75 % случаев, при этом вероятно, что мутации в других пока неидентифицированных генах могут также вызывать развитие ПБЭ .
При ПоБЭ чаще всего мутации обнаруживаются в генах LAMB3 (70 % случаев), LAMA3, LAMC2, COL17A1. В большинстве случаев заболевание наследуется по аутосомно-рецессивному типу, однако, описаны случаи соматического мозаицизма и однородительской дисомии .
При ДБЭ описаны мутации в гене COL7A1, в 95 % случаев доминантного и рецессивного типов БЭ .
Обратный мозаицизм при БЭ
Обратный мозаицизм известен при различных наследственных болезнях. При обратном мозаицизме от родителей наследуется мутация, вызывающая заболевание, но затем, в результате определенных генетических событий, происходит полное или частичное восстановление функции мутированного гена (такое восстановление называется реверсией, а клетки, в которых оно произошло, называются ревертантными). Обратной трансформации могут подвергаться клетки разных типов: гепатоциты, лимфоциты, а в случае с БЭ — кератиноциты.
Впервые обратный мозаицизм при БЭ был описан Джонкменом и соавторами у больной пограничным БЭ подтипа не-Херлитца. Явление обратного мозаицизма при БЭ легло в основу разработки радикальных методов лечения .
Клинические проявления буллёзного эпидермолиза
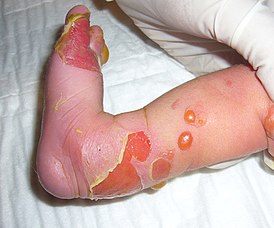
Отличительным признаком БЭ является ранимость кожи, её чувствительность к любому механическому воздействию. Это неизбежно связано с возникновением эрозий. При большинстве форм БЭ эрозиям предшествуют напряженные пузыри, большая часть которых наполнена прозрачной бесцветной жидкостью, иногда пузыри имеют геморрагическое содержимое .
Простой буллёзный эпидермолиз

Современная классификация буллёзного эпидермолиза делит ПБЭ на 12 подтипов. Наиболее распространёнными подтипами ПБЭ являются: локализованный подтип (ранее тип Вебера-Коккейна); генерализованный подтип (ранее Доулинга-Меары или герпетиформный), генерализованный подтип другой (ранее Кёбнера), простой буллёзный эпидермолиз с пятнистой пигментацией.
Фенотип данных подтипов варьируется, пузыри могут появляться на кистях рук и стопах, а могут покрывать все тело, обычно пузыри заживают без образования рубцов. В редких случаях наличие множественных распространенных пузырей приводит к летальному исходу в случае присоединения вторичной инфекции .
Наиболее распространённым подтипом простого БЭ является локализованный подтип, обычно в семьях большое количество больных и заболевание встречается в нескольких поколениях. При данном подтипе пузыри локализуется на ладонях и подошвах, однако в раннем возрасте могут носить распространённый характер, с возрастом проявления минимизируются. Обострение клинических проявлений происходит в летнее время .
Самый тяжёлый вариант простого БЭ является генерализованный подтип Доулинга-Меары. Он характеризуется наличием пузырей или везикул, возникающих группами (отсюда название «герпетиформный простой БЭ», поскольку некоторые повреждения могут имитировать повреждения, возникающие при простом герпесе). Заболевание проявляется в момент рождения, степень тяжести в значительной степени варьируется как в пределах одной семьи, так и по сравнению с другими семьями. При данном подтипе отмечается распространённый или сливной ладонно-подошвенный гиперкератоз, дистрофия ногтей, атрофическое рубцевание, милии, гипер- и гипопигментация и повреждение слизистых. Образование пузырей может принимать тяжёлую форму, иногда приводящую к смерти новорождённого или младенца. При данном подтипе может также возникать задержка роста, стеноз гортани и возможна преждевременная смерть .
Пограничный буллёзный эпидермолиз

Пограничный БЭ также характеризуется хрупкостью кожи и слизистых оболочек, спонтанным появлением пузырей, практически без травм. Одним из признаков является образование грануляционной ткани на определенных частях тела. Пузыри обычно заживают без существенных рубцов. В настоящее время пограничный тип БЭ делят на 2 основных подтипа, один из которых, подразделяется ещё на 6 подтипов. Основные подтипы пограничного БЭ: подтип Херлитца (ранее летальный); подтип не-Херлитца (ранее генерализованный атрофический) .
Подтип Херлитца наиболее тяжелый генерализованный вариант пограничного БЭ; при данном типе БЭ высокий риск преждевременной смерти. К типичным симптомам относятся образование множества пузырей, эрозий и атрофических рубцов кожи, ониходистрофия, приводящая к полной утрате ногтевых пластин и серьезным рубцам ногтевых лож, милии, тяжелое поражение мягких тканей в ротовой полости, гипоплазия эмали и тяжелый кариес зубов. Патогномоничным симптомом является обильная грануляционная ткань , симметрично образующаяся вокруг рта, в области средней части лица и вокруг носа, в верхней части спины, подмышечных впадинах и ногтевых валиках. Возможными системными осложнениями являются тяжёлая полиэтиологическая анемия, задержка роста, эрозии и стриктуры желудочно-кишечного тракта и поражение слизистых оболочек верхних дыхательных путей и мочеполового тракта, поражение почек, наружных оболочек глаза и, в редких случаях, поражение кистей рук. Смертность крайне высока, особенно в первые несколько лет жизни, в результате прекращения прибавки в весе, сепсиса, пневмонии или обструкции гортани и трахеи .
Подтип не-Херлитца проявляется образованием генерализованных пузырей, эрозий и корок на коже, атрофических рубцов, рубцовой алопеции («по мужскому типу»), дистрофией или потерей ногтей, гипоплазией эмали и кариесом.
Дистрофический буллёзный эпидермолиз

Дистрофический БЭ делится на два главных подтипа по типу наследования: доминантный и рецессивный дистрофический БЭ (ДДБЭ и РДБЭ).
Доминантный дистрофический БЭ клинически характеризуется рецидивирующим образованием пузырей, милиями и атрофическим рубцеванием, особенно на конечностях, а также дистрофией и, в конечном итоге, утратой ногтей. У большинства пациентов поражение кожи является генерализованным. Внекожные проявления характеризуются осложнениями со стороны желудочно-кишечного тракта.
Рецессивный дистрофический БЭ делится на 2 подтипа — тяжёлый генерализованный подтип (ранее Аллопо-Сименса) и генерализованный другой подтип (ранее не-Аллопо-Сименса). Рецессивный дистрофический тяжелый генерализованный подтип (ранее Аллопо-Сименса) характеризуется генерализованным образованием пузырей, эрозиями, атрофическими рубцами, ониходистрофией и утратой ногтей, псевдосиндактилией пальцев рук и ног. Поражение кожи носит обширный и устойчивый к терапии характер. Рецессивный дистрофический генерализованный другой подтип (ранее не-Аллопо-Сименса) характеризуется локализацией пузырей на руках, ногах, коленях и локтях, иногда на сгибах, на туловище.
При всех подтипах РДБЭ с возрастом развиваются контрактуры суставов локтей и коленей, кистей и стоп. Часто встречаются внекожные проявления, включающие поражения желудочно-кишечного и мочеполового трактов, внешних оболочек глаза, хроническую анемию, остеопороз, задержку роста. У пациентов с РДБЭ высокий риск онкологических заболеваний, в частности образования агрессивных плоскоклеточных карцином .
Лабораторная диагностика
Наиболее достоверным и надёжным методом установления диагноза является исследование образцов кожи, взятых при биопсии, с помощью трансмиссионной электронной микроскопии, однако сейчас основным методом диагностики БЭ стала непрямая иммунофлюоресценция, когда стали доступны моноклональные и поликлональные антитела против важнейших белков эпидермиса и границы эпидермиса и дермы, задействованных в патогенезе БЭ .
При БЭ с помощью иммуногистологических (иммуногистохимия и иммунофлюорисценция) методов определяется наличие, отсутствие или сниженная экспрессия структурных белков кератиноцитов или базальной мембраны, а также распределение тех или иных белков в естественно образующихся или искусственно вызванных пузырях . Тем самым можно определить уровень образования пузырей: внутри эпидермиса, на границе эпидермиса и дермы. Иммуногистологические методы, наряду с методами ДНК-диагностики, служат основными способами диагностики БЭ. С их помощью стало возможно быстро классифицировать БЭ и определить основной тип БЭ, определить структурный белок, наиболее вероятно подвергшийся мутации и определить клинический прогноз .
Результаты иммуногистологического исследования предоставляют основу для дальнейшего поиска мутаций, указывая на то, какие гены, кодирующие структурные белки кератиноцитов или базальной мембраны, необходимо исследовать.
Трансмиссионная электронная микроскопия позволяет осуществить визуализацию и полуколичественную оценку определенных структур кожи (нитей кератина, десмосом, гемидесмосом, якорных нитей, якорных волокон), о которых известно, что их количество и/или вид изменяется при определенных подтипах БЭ .
После определения типа или подтипа БЭ с помощью иммуногистологических методов возможно проведение генетического анализа. Генетическая диагностика позволяет выявить мутации, определить тип и локализацию мутации, а в итоге тип наследования заболевания. В настоящее время генетический анализ является методом, достоверно подтверждающим диагноз. При БЭ обычно генетический анализ проводят методом прямого секвенирования. Генетический анализ позволяет провести дородовую диагностику последующего потомства в семье, где есть пациент с БЭ .
Лечение
В настоящее время не существует радикальных методов лечения ни одной из форм БЭ. Несмотря на это, существуют эффективные превентивные и симптоматические методы терапии. В целом лечение должно быть комплексным и осуществляться мультидисциплинарной командой врачей, так как БЭ — это системное заболевание .
Лечение БЭ симптоматическое. Основным в лечении БЭ является уход за ранами с целью их быстрого заживления и эпителизации кожных покровов. Основной задачей является предотвратить разрастание размеров пузыря и повреждение его покрова с целью лучшего заживления раны и профилактики эрозии. Практикуется покрытие ран атравматичными неприлипающими материалами и многослойное бинтование .
Лечение любых осложнений также носит симптоматический характер и направлено на поддержание работы всех органов и систем. Пациенты с тяжелыми подтипами БЭ часто нуждаются в хирургическом вмешательстве на желудочно-кишечном тракте и в операциях по разделению пальцев .
8 ноября 2017 года университетская клиника города Бохум заявила об успешном окончании лечения методом трансплантации кожи, которое длилось два года . Ребенку, больному БЭ, удалось заменить практически всю кожу на здоровую трансгенную кожу, выращенную из его же клеток, в которые была вставлена работающая версия гена LAMB3.
Другие препараты: , .
Исследования
В настоящее время ученые ведут исследования по лечению БЭ в трёх направлениях: генная терапия, протеиновая (белковая) терапия и клеточная терапия (использование стволовых клеток). Все эти виды лечения находятся на разных этапах разработки. Изучением и лечением буллёзного эпидермолиза во всём мире занимается Международная ассоциация DEBRA International , которая была основана в 1978 г. в Великобритании. Ассоциация насчитывает более 40 стран-участниц, в каждой из которых действуют национальные научно-медицинские центры изучения БЭ. Представителем ассоциации в России является « Дети-бабочки ».
В 2015 году немецкие специалисты заменили 80 % кожи больного БЭ на трансгенную кожу
Примечания
- Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- [Fine JD., Hintner H. Life with Epidermolysis Bullosa (EB): Etiology, Diagnosis, Multidisciplinary Care and Therapy. 2009.]
- Дата обращения: 3 октября 2017. 4 октября 2018 года.
- Дата обращения: 3 октября 2017. 19 марта 2017 года.
- Дата обращения: 3 октября 2017. 5 декабря 2017 года.
- [Буллезный эпидермолиз. Под ред. Дж-Д.Файна, Х.Хинтнера. Пер. с англ. Под ред. Ю. Ю. Коталевской. Практика. Москва, 2014. С. 358.]
- . Дата обращения: 3 октября 2017. 11 октября 2018 года.
- [Koebner H (1886) Hereditre Anlage zur Blasenbildung (epidermolysis bullosa hereditaria). Dtsch Med Wochenschr 12:21-22.]
- Дата обращения: 3 октября 2017. 23 июля 2018 года.
- [Fine JD, Johnson LB, Suchindran C, et al (1999) The National Epidermolysis Bullosa Registry: organization, goals, methodologic approaches, basic demography, and accomplishments. In: Fine JD, Bauer EA, McGuireJ, Moshell A (eds) Epidermolysis bullosa: clinical, epidemiologic, and laboratory advances, and the findings of the National Epidermolysis Bullosa Registry. Johns Hopkins University Press, Baltimore.]
- [Fine JD, Johnson LB, Suchindran C, et al (1999) The epidemiology of inherited EB: findings within American, Canadian, and European study populations. In: Fine JD, Bauer EA, McGuire J, Moshell A (eds) Epidermolysis bullosa: clinical, epidemiologic, and laboratory advances, and the findings of the National Epidermolysis Bullosa Registry. Johns Hopkins University Press, Baltimore.]
- Дата обращения: 3 октября 2017. 19 сентября 2016 года.
- Дата обращения: 3 октября 2017. 19 сентября 2016 года.
- Дата обращения: 3 октября 2017. 20 сентября 2016 года.
- Дата обращения: 3 октября 2017. 19 сентября 2016 года.
- Дата обращения: 3 октября 2017. 19 сентября 2016 года.
- Дата обращения: 3 октября 2017. 19 сентября 2016 года.
- Дата обращения: 3 октября 2017. 19 марта 2017 года.
- Дата обращения: 3 октября 2017. 19 сентября 2016 года.
- Дата обращения: 3 октября 2017. 19 сентября 2016 года.
- Дата обращения: 3 октября 2017. 19 сентября 2016 года.
- Дата обращения: 3 октября 2017. 23 сентября 2016 года.
- Дата обращения: 3 октября 2017. 20 сентября 2016 года.
- Дата обращения: 3 октября 2017. 21 сентября 2016 года.
- Дата обращения: 3 октября 2017. 21 сентября 2016 года.
- Дата обращения: 3 октября 2017. 20 сентября 2016 года.
- Дата обращения: 3 октября 2017. 22 сентября 2016 года.
- [Gedde-Dahl T Jr (1971) Epidermolysis bullosa. A clinical, genetic and epidemiologic study. The Johns Hopkins Press, Baltimore.]
- Дата обращения: 3 октября 2017. 19 сентября 2016 года.
- Дата обращения: 3 октября 2017. 23 сентября 2016 года.
- Дата обращения: 3 октября 2017. 23 сентября 2016 года.
- Дата обращения: 3 октября 2017. 8 ноября 2017 года.
- Дата обращения: 3 октября 2017. 19 сентября 2016 года.
- Дата обращения: 3 октября 2017. 20 сентября 2016 года.
- Дата обращения: 20 июля 2015. Архивировано из 5 марта 2016 года.
- Дата обращения: 20 июля 2015. Архивировано из 17 июня 2016 года.
- Дата обращения: 3 октября 2017. 19 сентября 2016 года.
- Дата обращения: 3 октября 2017. 20 сентября 2016 года.
- Fine JD, Johnson LB, Suchindran C, et al (1999) Cancer and inherited epidermolysis bullosa: lifetable analyses of the National Epidermolysis Bullosa Registry study population. In: Fine JD, Bauer EA, McGuire J, Moshell A (eds) Epidermolysis bullosa: clinical, epidemiologic, and laboratory advances, and the findings of the National Epidermolysis Bullosa Registry. Johns Hopkins University Press, Baltimore.
- Дата обращения: 3 октября 2017. 23 сентября 2016 года.
- Дата обращения: 3 октября 2017. 20 сентября 2016 года.
- Дата обращения: 3 октября 2017. 20 сентября 2016 года.
- [Eady RAJ (1998) Epidermolysis bullosa. In: Champion RH, Burton JL, Burns DA, Breathnach SM (eds) Textbook of Dermatology, 6th edn. Blackwell, Oxford.]
- [Uitto J (2005) Epidermolysis bullosa. In: Spitz JL (ed) Genodermatoses — A clinical guide to genetic skin disorders, 2nd edn. Lippincott Williams & Wilkins, Philadelphia.]
- Дата обращения: 3 октября 2017. 23 сентября 2016 года.
- Дата обращения: 3 октября 2017. 20 сентября 2016 года.
- Дата обращения: 3 октября 2017. 20 сентября 2016 года.
- Дата обращения: 3 октября 2017. 16 июля 2017 года.
- Дата обращения: 3 октября 2017. 3 апреля 2016 года.
- Дата обращения: 13 марта 2019. 25 марта 2019 года.
- Дата обращения: 13 марта 2019. 16 февраля 2019 года.
- Дата обращения: 13 марта 2019. 20 августа 2019 года.
- Дата обращения: 13 марта 2019. 10 ноября 2017 года.
- . Дата обращения: 20 июля 2015. Архивировано из 29 июля 2015 года.
- Анна Казнадзей. . nplus1.ru. Дата обращения: 9 ноября 2017. 9 ноября 2017 года.
Ссылки
- 2020-09-20
- 1

