Альтернирующие синдромы
- 1 year ago
- 0
- 0
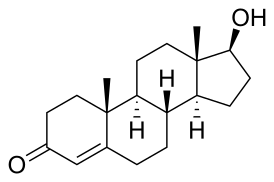
Синдро́м Ка́ллмана ( Ка́льмана ) — симптомокомплекс наследственно обусловленных аномалий, характеризующийся сочетанием гипогонадотропного гипогонадизма с расстройствами обоняния ( аносмия или гипоосмия) и недостаточной секрецией гонадотропин-рилизинг гормона (ГнРГ). Низкий уровень ЛГ и ФСГ ведёт к развитию вторичного (центрального) гипогонадизма .
В 1944 году и соавт. в работе «Генетические аспекты первичного евнухоидизма» впервые описали синдром, характеризующийся задержкой или отсутствием полового развития и аносмией . Лишь после идентификации гонадолиберина было показано, что селективная недостаточность ЛГ и ФСГ (гонадотропинов гипофиза) является следствием изолированного дефицита секреции именно этого гормона.
Причина аномалий при синдроме Каллмана — расстройство импульсной секреции гонадолиберина (ГнРГ) в гипоталамусе . Наследование X-сцепленное рецессивное или аутосомно-доминантное с неполной пенетрантностью. У пациентов выявлены дефекты гена, контролирующего миграцию гонадолиберинсекретирующих нейронов в и впоследствии в гипоталамус, однако дефекты гена, кодирующего гонадолиберин не обнаружены .
Недостаток гонадотропин-рилизинг гормона связан с делецией KAL-гена, локализованного на коротком плече X-хромосомы , который кодирует экстрацеллюлярный матрикс гликопротеина (аносмина-1), относящегося к классу нейромолекул, обладающих адгезивными свойствами, позволяющими ГнРГ-секретирующим нейронам входить в обонятельную луковицу и мигрировать в гипоталамус в процессе эмбриогенеза . Низкий уровень ЛГ и ФСГ обусловливает вторичный гипогонадизм .
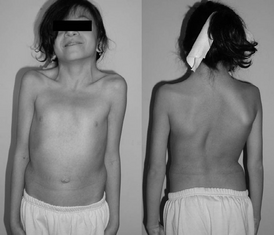
Основные компоненты: вторичный (центральный) гипогонадизм и аносмия (или гипоосмия). У части пациентов отмечается цветовая слепота и тяжёлые аномалии головного мозга и черепа по средней линии .
Характерен евнухоидизм , нередко наблюдается крипторхизм . Встречается соматическая патология: расщепление верхней губы (« заячья губа ») и твёрдого нёба (« волчья пасть »), полидактилия ( шестипалость ), высокое «готическое» нёбо, укорочение уздечки языка, асимметрия лица, гинекомастия , функциональные нарушения сердечно-сосудистой системы. Так как заболевание характеризуется выпаданием лишь одного , все остальные гормоны гипоталамуса секретируются нормально, в том числе соматолиберин . Из-за этого развитие и рост организма, хоть и с запаздыванием, происходит нормально с поправкой на то, что у организма не развиваются половые железы. Из-за этого несоответствия зоны роста в эпифизарных пластинках не окостеневают из-за отсутствия половых гормонов. Это в итоге приводит к высокорослости и относительно длинным конечностям по отношению к длине туловища. Этот механизм обусловливает евнухоидизм . Подобные пороки развития можно выявить и среди членов семьи пациента. При рентгенологическом исследовании кистей рук и лучезапястных суставов в прямой проекции обнаруживается запаздывание сроков окостенения (возрастной дифференцировки) .
У мальчиков в зависимости от степени выраженности недостаточности гонадолиберина выявляется разной степени выраженности половой инфантилизм вплоть до полного отсутствия признаков пубертата, отсутствия вторичных половых признаков, небольших размеров тестикулы и азооспермия . Признаки вторичного гипогонадизма могут сочетаться с нарушением цветного зрения, атрофией зрительного нерва , глухонемотой, подковообразной почкой и крипторхизмом . У девочек — первичной аменореей .
В отличие от гипопитуитаризма , при синдроме Каллмана после многократного введения гонадолиберина уровни ЛГ и ФСГ повышаются.
Заместительная гормональная терапия — используют гонадотропины, дозы подбирают индивидуально .
Для жизни — благоприятный. При систематической правильно подобранной терапии развиваются вторичные половые признаки, появляется либидо , эрекции , поллюции . Пациенты способны вступать в брак, в некоторых случаях восстанавливается и репродуктивная функция .