Interested Article - Болезнь Вольмана

- 2021-04-01
- 1
Болезнь Вольмана ( синдром Вольмана , дефицит лизосомной кислой липазы ) — (LAL-D, ДЛКЛ) – это редкое аутосомно-рецессивное заболевание лизосомального накопления , вызванное повреждающими мутациями гена LIPA. Возраст начала заболевания и темпы его прогрессирования в значительной степени вариабельны и могут быть связаны с природой, лежащих в основе мутаций . Заболевание у пациентов грудного возраста имеет наиболее быстро прогрессирующее течение с развитием проявлений и симптомов в первые недели жизни; эти пациенты редко доживают до возраста, превышающего 6 месяцев. У детей старшего возраста и взрослых заболевание обычно характеризуется определенным сочетанием дислипидемии , гепатомегалии , повышением уровня трансаминаз и микровезикулярным стеатозом в биопсийном материале. У большей части пациентов наблюдается повреждение печени с исходом в фиброз , цирроз и печеночную недостаточность . Частыми изменениями являются повышение уровней холестерина липопротеинов низкой плотности и снижение уровней холестерина липопротеинов высокой плотности. Начиная с детского возраста, могут проявляться и нарушения со стороны сердечно-сосудистой системы . Учитывая, что эти клинические проявления могут наблюдаться и при других сердечно-сосудистых нарушениях, заболеваниях печени и метаболических расстройствах, неудивительно, что LAL-D часто не диагностируется в клинической практике .
Эпоним
В 1956 году Абрамов ( A. Abramov ), Шорр ( S. Schorr ) и Вольман ( M. Wolman ) описали данное наследственное заболевание у ребёнка, который родился от близкородственного брака. Болезнь получила название в честь невролога (1914—2009) .
Эпидемиология
Болезнь Вольмана является редким наследственным заболеванием — распространённость по разным источникам может колебаться от 1:40000 до 1:300000. Исследование Scott определяет частоту 1:130 000 .
Патогенез
В норме нейтральные жиры (эфиры холестерина и, в меньшей степени, триглицериды ), Попавшие в лизосому путём рецептор-опосредованного эндоцитоза , под воздействием ЛКЛ распадаются до свободного холестерина и жирных кислот . Эти липиды и их окисленные производные вступают во взаимодействие с факторами транскрипции (стериновым регуляторным элементом связывания белков [СРЭСБ]), которые непосредственно модулируют экспрессию генов, вовлечённых в синтез и захват холестерина , а также липогенез .
При отсутствии или снижении активности ЛКЛ эфиры холестерина и триглицериды не распадаются и накапливаются в лизосомах. Недостаток свободного холестерина в клетке приводит к СРЭСБ-опосредованной стимуляции эндогенного синтеза холестерина, ингибированию гидроксиметилглутарил-коэнзим А (ГМГ-КоА) редуктазой и эндоцитоза посредством рецепторов ЛПНП . Параллельно с этим увеличивается синтез аполипопротеина В (АпоВ) и значительно повышается образование холестерина, липопротеинов очень низкой плотности (ЛПОНП). Увеличение экспрессии ГМГ-КоА редуктазы является первичным результатом СРЭСБ -2-опосредованного внутриклеточного уменьшения холестерина, приводя к увеличению уровня свободного холестерина.
Влияние какого-либо увеличения уровня свободного холестерина, обусловленного стимулированием ГМГ-КоА редуктазы при ЛКЛ-D, изучено недостаточно, однако это может вызвать ингибирование активности рецепторов ЛПНП по механизму обратной связи с уменьшением клиренса холестерина ЛПНП из кровотока.
У пациентов с ДЛКЛ, обычно, имеет место дислипидемия с повышенным уровнем общего холестерина в сыворотке крови, высоким уровнем холестерина ЛПНП, низким уровнем холестерина липопротеинов высокой плотности (ЛПВП), а также возможным повышением уровня триглицеридов. Повышение уровня общего холестерина и триглицеридов связано с накоплением в плазме крови АпоВ-содержащих липопротеинов, таких как холестерин ЛПОНП и холестерин ЛПНП.
При ДЛКЛ накопление эфиров холестерина в лизосомах и, как следствие, уменьшение внутриклеточного содержания свободного холестерина приводит к: 1) снижению образования оксистерина и последующему подавлению активации экспрессии аденозинтрифосфат-связывающегося кассетного транспортера A1 (ABCA1). ABCA1; и 2) уменьшению количества холестерина в субклеточных образованиях и в плазматической мембране, доступного для ABCA1 -опосредованного переноса на внеклеточный обедненный липидами аполипопротеин A1. Данное явление является ключевым в формировании частиц α-ЛПВП .
Наследование
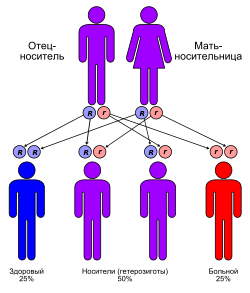
Болезнь Вольмана наследуется, как и подавляющее большинство лизосомных болезней накопления , по аутосомно-рецессивному типу наследования . Следовательно, с одинаковой частотой встречается как у мужчин , так и у женщин . Заболевание клинически манифестирует только в случае, когда обе аутосомы , полученные по одной от отца и матери, являются дефектными по гену LIPA (повреждение обеих копий гена, находящихся на гомологичных аутосомах 10q23.2-23.3) .
Классификация
Согласно Международной классификации болезней десятого пересмотра ( ), различают:
-
E
Нарушения обмена
сфинголипидов
и другие болезни накопления
липидов
.
- E Другие нарушения накопления липидов. Церебротендинозный холестероз ( ван Богарта—Шерера—Эпштейна ), Болезнь Вольмана .
Клиническая картина
Клинически дефицит лизосомной кислой липазы приводит к двум основным фенотипам: болезнь Вольмана (БВ), начинающаяся в младенческом возрасте (MIM 278000), и болезнь накопления эфиров холестерина (БНЭХ), начинающаяся в более позднем возрасте, которые были впервые описаны в 1956 г. и в 1963 г., соответственно. По мере увеличения возраста начала / диагностики заболевания его тяжесть снижается, а вариабельность фенотипических проявлений – возрастает.
Имеющиеся у пациентов проявления и симптомы дефицита кислой лизосомальной липазы (LAL-D, ДЛКЛ) могут значительно варьировать. Многие из наиболее распространенных клинических проявлений, в частности, дислипидемия, гепатомегалия и повреждение клеток печени (по данным повышения уровня трансаминаз в сыворотке крови с прогрессированием в фиброз и цирроз), совпадают с аналогичными проявлениями других сердечно-сосудистых заболеваний, заболеваний печени и метаболических расстройств, которые более распространены, чем ДЛКЛ.
У детей грудного возраста по сравнению с детьми старшего возраста и взрослыми ДЛКЛ, обычно, имеет более острое клиническое течение. В качестве первых регистрируемых проявлений часто выступают симптомы со стороны желудочно-кишечного тракта (рвота, диарея со стеатореей и вздутие живота), а также отставание в росте. Вздутие живота является отличительной чертой, обусловленной, в первую очередь, массивной гепатоспленомегалией. У приблизительно 50 % детей грудного возраста, а также у детей более старшего возраста при рентгенологическом исследовании в надпочечниках обнаруживаются характерные точечные очаги кальцификации. Также может наблюдаться анемия. Более поздние проявления связаны с полиорганной недостаточностью, в частности, с циррозом печени / печеночной недостаточностью, и включают в себя желтуху и кахексию [15]. Эффекты на центральную нервную систему наблюдаются нечасто, а выявляемые скорее связаны с недостаточным питанием и/или специфическим недостатком тех или иных питательных веществ, либо с осложнениями трансплантации костного мозга, чем непосредственно с ДЛКЛ .
Лечение
Заместительная ферментная терапия для лечения болезни Вольмана разработана фармацевтической компанией Alexion (США), специализирующейся на орфанных заболеваниях. Для лечения этого заболевания компания предлагает препарат Kanuma™ ( sebelipase alfa ), являющийся рекомбинантной формой кислой липазы человека. В настоящее время (зима 2016) препарат зарегистрирован в США, Европе, Японии, Канаде. На территории РФ зарегистрирован препарат Канума (Себелипаза альфа) производства Алексион Юроп САС
.
Прогноз
В зависимости от формы (времени начала заболевания). У детей грудного возраста быстро приводит к летальному исходу (в течение 12 месяцев). У детей и взрослых с ДЛКЛ на протяжении 3 лет после начала симптомов приблизительно в 50 % случаев наблюдается прогрессирование в фиброз, цирроз или возникает необходимость в проведении трансплантации печени . Введение в клиническую практику препарата себелипаза альфа (Канума) изменит ситуацию - все больные, получающие ферементозаместительную терапию возвращаются к нормальной жизни
См. также
- Недостаточность кислой фосфатазы
- Синдром ван Богарта — Шерера — Эпштейна
- Генные болезни
- Паренхиматозные дистрофии
- Лизосомные болезни накопления
Примечания
- Željko Reiner, Ornella Guardamagna, Devaki Nair, Handrean Soran, Kees Hovingh. // Atherosclerosis. — 2014-07-01. — Т. 235 , вып. 1 . — С. 21–30 . — ISSN . — doi : . 11 апреля 2016 года.
- (англ.) . Wolman's disease . whonamedit.com. Дата обращения: 9 января 2015. 9 января 2015 года.
- M. Wolman, V. V. Sterk, S. Gatt, M. Frenkel. (англ.) // Pediatrics, Evanston, Illinois . — 1961. — Vol. 28 . — P. 742-757 . — . 24 октября 2019 года.
- Stuart A. Scott, Benny Liu, Irina Nazarenko, Suparna Martis, Julia Kozlitina. // Hepatology (Baltimore, Md.). — 2013-09-01. — Т. 58 , вып. 3 . — С. 958–965 . — ISSN . — doi : . 11 января 2022 года.
- ↑ Grabowski GA, Charnas L, Du H. (англ.) // Metab Mol Bases Inherit Dis . — 2012. — Vol. 142 . — P. 1-31 .
- (нем.) . Funktionelle Ultrastruktur. Verlag Springer, 2005, ISBN 978-3-211-83563-0 S. 110-111. . springerlink.com. Дата обращения: 9 января 2015. (недоступная ссылка)
- . Глава 316. Лизосомные болезни накопления (с. 250—273) . med-books.info. Дата обращения: 9 января 2015. 7 июня 2015 года.
- Anderson Richard A. , Rao Nagesh , Byrum Robert S. , Rothschild Cynthia B. , Bowden Donald W. , Hayworth Rosa , Pettenati Mark. (англ.) // Genomics. — 1993. — January ( vol. 15 , no. 1 ). — P. 245—247 . — ISSN . — doi : . — .
- . grls.rosminzdrav.ru . Дата обращения: 27 апреля 2022. 27 апреля 2022 года.
- Bernstein DL, Hülkova H, Bialer MG, et al. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. // J Hepatol. — 2013. — Т. 58 , № 6 . — С. 1230-1243 . — doi : . — .
Ссылки
- (нем.)
- 2021-04-01
- 1

