Меркль, Вилли
- 1 year ago
- 0
- 0

Синдром Прадéра — Ви́лли — редкое наследственное заболевание , причиной которого является отсутствие отцовской копии участка хромосомы 15q11-13. В этом участке хромосомы 15 находятся гены , в регуляции которых задействован геномный импринтинг . Большинство случаев являются спорадическими, для редких описанных семейных случаев характерно неменделевское наследование .
Синдром впервые описали в 1956 году учёные из Швейцарии А. Прадер ( A. Prader ), Х. Вилли ( H. Willi ) и А. Лабхарт ( A. Labhart ) .
Частота встречаемости — 1 : 12 000-15 000 живорождённых младенцев. Патология встречается с одинаковой частотой у женщин и мужчин .
Наиболее частой причиной синдрома (70-75 % случаев) является делеция участка 15q11-13 хромосомы 15, унаследованной от отца. Около четверти случаев обусловлено однородительской дисомией хромосомы 15 upd(15)mat , когда обе 15-е хромосомы у пациента являются копиями материнского происхождения. В незначительном числе случаев синдром связан с нарушением импринтинга или с наличием сбалансированной транслокации с точкой разрыва внутри участка 15q11-13 .
Для синдрома Прадера — Вилли характерны:
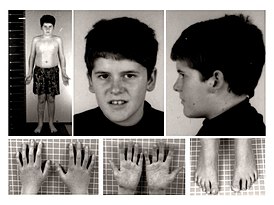
Внешние признаки: у взрослых выражена переносица, лоб высокий и узкий, глаза, как правило, миндалевидные, губы тонкие.
Как правило, у больного встречается не более пяти вышеуказанных признаков.

Синдром Прадера — Вилли (СПВ) связан с эпигенетическим феноменом, известным как импринтинг . Обычно плод наследует импринтированную материнскую копию генов PW и функциональную отцовскую копию генов PW. Из-за импринтинга копии этих генов, унаследованные от матери, практически молчат, и поэтому плод развивается благодаря экспрессии отцовских копий генов . Однако при СПВ происходит мутация/делеция отцовских копий генов PW, в результате чего плод остается без функционирующих генов PW. Гены PW представляют собой гены и , а также кластеры snoRNA : , SNORD107, SNORD108 и две копии SNORD109, 29 копий (HBII-85) и 48 копий (HBII-52). Эти гены расположены в области 15q11-13 хромосомы 15 . Эта так называемая область PWS/AS в отцовской хромосоме 15 может быть утрачена по одному из нескольких генетических механизмов, что в большинстве случаев происходит в результате случайной мутации. Другие, менее распространенные механизмы включают однородительскую дисомию , спорадические мутации , хромосомные транслокации и делеции генов.
Регион 15q11-13 вовлечен как в синдром Прадера — Вилли, так и в синдром Ангельмана . В то время как СПВ возникает в результате потери генов PW в этом регионе отцовской хромосомы, потеря другого гена ( ) в том же регионе материнской хромосомы вызывает синдром Ангельмана . СПВ и синдром Ангельмана представляют собой первые зарегистрированные случаи нарушений, связанных с импринтингом, у людей.
Риск развития СПВ для брата или сестры больного ребенка зависит от генетического механизма, вызвавшего заболевание. Риск для братьев и сестер составляет <1%, если у больного ребенка имеется делеция гена или однородительская дисомия, до 50%, если у больного ребенка есть мутация контрольной области импринтинга, и до 25%, если присутствует родительская хромосомная транслокация. Пренатальная диагностика возможна для любого из известных генетических механизмов.
Микро делеция в одном семействе мякРНК HBII-52 исключила её важную роль в заболевании .
Исследования модельных систем человека и мыши показали, что делеция 29 копий мякРНК C/D-бокса SNORD116 (HBII-85) является основной причиной СПВ .
Синдром диагностируется путём генетического анализа, рекомендуемого для новорождённых с пониженным мышечным тонусом (гипотонусом). Иногда вместо диагноза «синдром Прадера — Вилли» врачи ошибочно ставят диагноз « синдром Дауна » (поскольку синдром Дауна встречается намного чаще).
Дети с синдромом Прадера — Вилли очень похожи между собой, опытный генетик сможет быть уверен в диагнозе, не дожидаясь результатов исследования кариотипа.
Синдром Прадера — Вилли является врождённой генетической аномалией; в настоящее время специфические способы его лечения не разработаны. Однако некоторые лечебные мероприятия повышают качество жизни людей с синдромом. В частности, младенцы с гипотонусом должны получать массаж и другие виды специальной терапии. Рекомендуются использование специальных методик развития ребёнка, занятия с логопедом и дефектологом . Показан приём « гормонов роста», заместительная гормональная терапия (с применением гонадотропинов ).
Гипогонадизм обычно проявляется в микропении ( микропенис — половой член аномально малого размера) и неопущении яичек у мальчиков ( крипторхизм ); врачи могут посоветовать подождать, пока яички опустятся сами, или порекомендовать хирургическое вмешательство либо гормонотерапию.
Для коррекции избыточной массы тела применяется диета с ограничением количества жиров и углеводов . Из-за ожирения, сопутствующего синдрому, нужно пристально следить за количеством и качеством пищи, поглощаемой человеком с синдромом Прадера — Вилли (обычно люди с таким синдромом способны много съесть, не насыщаясь).
Возможным осложнением может стать апноэ (задержка дыхания во сне).
Риск, что следующий ребёнок у тех же родителей родится также с синдромом Прадера — Вилли, зависит от механизма, вызвавшего генетический сбой.
Этот риск меньше 1 %, если у первого ребёнка делеция гена или партеногенетическая (однородительская) дисомия; составляет до 50 %, если сбой вызван мутацией; до 25 % — в случае транслокации родительских хромосом. Родителям рекомендуется пройти генетическое обследование.
У большинства людей с синдромом Прадера — Вилли наблюдается задержка психического и речевого развития. Согласно исследованиям, которые провели Л. М. Керфс и Дж. П. Фринс (1992) ,
По другим исследованиям (Кэссиди), 40 % пациентов с синдромом Прадера — Вилли демонстрируют интеллект на грани среднего или сниженный уровень интеллекта.
Как правило, дети с синдромом Прадера — Вилли имеют хорошую долговременную зрительную память, они могут научиться читать, могут обладать богатым пассивным словарём, но их собственная речь обычно хуже, чем понимание. Слуховая память, математические навыки и навыки письма, зрительная и слуховая кратковременная память у таких детей обычно значительно хуже.
Синдром Прадера — Вилли нередко ассоциируется с повышенным аппетитом. У больных повышена концентрация в крови гормона грелина . Для них также характерна пониженная концентрация соматолиберина . Это обусловлено тем, что 15-я хромосома связана с гипоталамусом . Однако при вскрытии умерших с синдромом Прадера — Вилли не было обнаружено никаких дефектов гипоталамуса. По другим данным, наблюдалось снижение общего количества клеток и окситоцин -содержащих клеток паравентрикулярных ядер гипоталамуса
| Аутосомные |
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| X / Y связанные |
|
||||||||
| Транслокации |
|
||||||||
| Иные | |||||||||
| Пороки ЦНС | |
|---|---|
|
Синдромы,
с нарушением развития ЦНС |
|