Патологическая анатомия
- 1 year ago
- 0
- 0

Первичные иммунодефициты — наследственные или приобретённые во внутриутробном периоде иммунодефицитные состояния. Обычно они проявляются или сразу после рождения, или в течение первых двух лет жизни (врождённые иммунодефициты). Однако менее выраженные генетические дефекты иммунного ответа могут манифестировать позже, например, на втором—третьем десятилетии жизни (например, общая вариабельная иммунная недостаточность). Наследственные формы первичных иммунодефицитов, как правило, характеризуются аутосомно-рецессивным или Х-сцепленным рецессивным типом наследования. Первым в 1952 г. был описан синдром Бру́тона .
Согласно последней классификации международного союза иммунологических сообществ (IUIS) комитетом по врожденным ошибкам иммунитета все ПИД разделяются на 9 классов :
К первичным дефицитам клеточного иммунитета относятся следующие заболевания:
В основе синдрома Ди Джо́рджи ( Di George ) лежит гипоплазия тимуса. Синдром описан в 1965 г. Считается, что это заболевание не является наследственным, оно возникает в результате приобретённого нарушения органогенеза в области III—V жаберных дуг (глоточных карманов) на 6—8 неделе беременности. Поэтому, кроме порока тимуса, отмечаются дефекты околощитовидных желёз, сердца и крупных сосудов, а также орофациальные пороки (микростомия, микрогнатия, гипертелоризм, низкое расположение ушных раковин).
Результатом гипоплазии паращитовидных желёз является дефицит парат-гормона и персистирующая гипокальциемия, вследствие чего развивается судорожный синдром, который может проявиться уже в первые часы жизни (неонатальная тетания). Причиной смерти детей в более старшем возрасте служат осложнения, связанные с пороками развития сердца.
Нарушения, затрагивающие Т-лимфоциты, могут быть как очень глубокими, так и едва заметными. В любом случае функция Т-клеток с возрастом восстанавливается и к 5 годам, если ребёнок остаётся жив, не удаётся обнаружить их недостаточности. Антиген-независимый этап созревания Т-клеток при этом происходит вне тимуса — в многослойных плоских эпителиях, прежде всего в эпидермисе. Одним из эффективных способов лечения синдрома Ди Джорджи является трансплантация эмбриональной ткани тимуса.
Синдром Ду́нкана (Х-сцепленный лимфопролиферативный синдром) — иммунодефицит, характеризующийся повышенной чувствительностью к вирусу Эпштейна — Барр . Ген повышенной чувствительности к вирусу локализован в Х-хромосоме, тип наследования заболевания рецессивный, поэтому болеют мальчики. У больных, перенёсших инфекционный мононуклеоз , развиваются длительное лихорадочное состояние, лимфаденопатия (увеличение лимфатических узлов), лимфоцитоз периферической крови, гепато- и спленомегалия. Позднее формируется В-клеточная лимфома, чаще в терминальных отделах тонкой кишки, от которой больные и погибают. Летальные исходы обусловлены также деструктивным гепатитом, вызываемым вирусом Эпштейна — Барр.
Недостаточность пурин-нуклеозид-фосфорилазы (ПНФ) наследуется по аутосомно-рецессивному типу. Дети страдают гипопластической анемией и крайне сниженной функцией Т-клеток.
Оротацидури́я — наследственное заболевание синтеза пиримидинов, которое проявляется повышенной экскрецией оротовой кислоты ( оротата ) с мочой, недостаточностью Т-лимфоцитов, мегалобластной анемией и задержкой умственного и физического развития. При этом заболевании снижена активность ферментов оротидил-пирофосфорилазы и оротидил-декарбоксилазы , которые преобразуют оротовую кислоту в нуклеотид-оротидин-монофосфат , необходимый для синтеза нуклеиновых кислот.
Биотин-зависимые ферментопатии также сопровождаются развитием клеточного иммунодефицита (наследственные дефекты биотинидазы и биотин-зависимых энзимов пируват-карбоксилазы и пропионат-карбоксилазы , участвующих в метаболизме аминокислот с разветвлённой цепью — валина, лейцина, изолейцина). Заболевание проявляется уже в периоде новорождённости эпизодами кетоацидоза, неврологической симптоматикой, алопецией, кожными сыпями и непереносимостью белка (рвота, мальдигестия, дегидратация). В моче содержится большое количество органических кислот. Дети отстают в физическом развитии. Из инфекционных процессов наиболее часто развиваются кандидоз и кератоконъюнктивиты. Биотин даёт хороший терапевтический эффект.
К первичным дефицитам гуморального иммунитета относятся следующие основные синдромы:
Наследственные дефициты гуморального иммунитета, за исключением синдрома Брутона и позднего иммунного старта, объединяют общим термином дисгаммаглобулинемии . При некоторых формах дисгаммаглобулинемий определяется нормальный или даже повышенный уровень иммуноглобулинов в крови и секретах слизистых оболочек.
Синдром Бру́тона ( агаммаглобулинемия бру́тоновского типа ) — недостаточность иммуноглобулинов всех классов. Заболевание было первым изученным иммунодефицитом наследственной природы (O. G. Bruton, 1952 ). Тип наследования — рецессивный, сцепленный с Х-хромосомой. В первые годы жизни развиваются инфекционные осложнения, преимущественно бактериальные. В то же время вирусные инфекции протекают у больных, как правило, легко. Первые признаки иммунодефицита становятся заметны на втором году жизни, хотя рецидивирующие инфекции могут появиться и у 8-месячного грудного младенца, и у 3-летнего ребёнка. Примерно у трети больных развивается вялотекущий артрит, похожий на ревматоидный , со стерильным выпотом в полость одного из крупных суставов.
В том случае, если заместительная терапия (введение препаратов иммуноглобулинов) начата прежде, чем повторные инфекции вызовут серьёзные морфологические изменения (например, бронхоэктазы, хроническую пневмонию и дыхательную недостаточность), ближайший прогноз очень хороший. Однако в подростковом и юношеском возрасте нередко развивается постепенно прогрессирующее неврологическое заболевание, напоминающее медленную вирусную инфекцию и проявляющееся дерматомиозитоподобным синдромом с выраженными отёками и периваскулярными лимфогистиоплазмоцитарными инфильтратами. Это тяжёлое системное заболевание приводит к летальному исходу. Считается, что оно вызвано энтеровирусной инфекцией (из крови и ликвора больных и умерших неоднократно выделяли энтеровирусы). В целом необходимо отметить высокую чувствительность этих больных к энтеровирусам. Так, дети с синдромом Брутона чаще болеют полиомиелитом , и он протекает у них тяжелее.
Избирательная недостаточность иммуноглобулинов проявляется стойким дефицитом иммуноглобулинов одного или нескольких классов (прежде всего, IgА и IgG).
Синдром Ве́ста — недостаточность иммуноглобулинов класса А. Заболевание проявляется частыми инфекционными поражениями конъюнктивы, респираторного и желудочно-кишечного трактов.
Недостаточность секреторной фазы — первичный дефицит IgA кишечника.
Выявлены случаи наследственной недостаточности синтеза каппа-цепей иммуноглобулинов.
Избирательная недостаточность иммуноглобулина M
Основная статья Недостаточность иммуноглобулина M
Недостаточность транскобаламина II проявляется В12-ахрестической анемией с характерными признаками мегалобластной анемии в виде атрофии слизистых оболочек и фуникулярного миелоза (атрофические процессы в ткани спинного мозга, сопровождающиеся развитием парезов, параличей и нарушениями чувствительности). Кроме того, страдает терминальная дифференцировка В-лимфоцитов. Они не способны трансформироваться в плазматические клетки и синтезировать иммуноглобулины.
Гипер-IgМ-синдром — наследственное заболевание, при котором отмечается недостаточность IgA и IgG, но высокий уровень IgМ. Подобная дисфункция гуморального иммунитета обнаружена у взрослых, страдающих частыми инфекциями дыхательных путей с развитием бронхоэктазов, а также у детей с врождённой краснухой. У некоторых больных полностью отсутствуют γ- и α-плазмоциты, и тогда в организме вырабатываются только IgM.
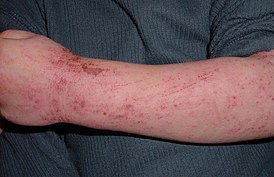
Гипер-IgЕ-синдром ( синдром Джоба , синдром золотистого стафилококка с гипер-IgЕ ) описан в 1966 г. Эпонимический термин включает фамилию больного. Мальчики болеют чаще (60—70 % случаев). В первые месяцы жизни возникают различные инфекционные процессы, вызываемые в основном золотистым стафилококком ( S. aureus ). В крови выявляется эозинофилия, иногда значительная, и нейтрофильный гранулоцитоз со сдвигом формулы влево. Несмотря на высокий уровень IgЕ и гистамина, проявлений анафилаксии и атопии не наблюдается. Среди IgЕ-антител преобладают антистафилококковые идиотипы. Нередко в подкожной клетчатке формируются холодные абсцессы .
Гипер-IgD-синдром с периодической лихорадкой ( синдром ван дер Ме́ера ). Заболевание описано в 1984 г. Оно проявляется рецидивирующими лихорадочными состояниями, лейкоцитозом до 10—20 тысяч клеток в мкл, головными болями, увеличением миндалин и значительным повышением концентрации IgD в крови.
Транзиторная недостаточность иммуноглобулинов в первые месяцы жизни. У новорождённого и грудного ребёнка в первые 3—4 мес. жизни отмечается низкая концентрация в крови иммуноглобулинов, особенно IgG. После рождения уровень IgG, обеспечиваемый пассивным проникновением через плаценту от матери, в течение первого месяца жизни стремительно падает, стабилизируется на втором, а затем начинает расти. Поскольку эта особенность выявляется практически у каждого ребёнка, она не считается патологической, а расценивается как пограничное транзиторное состояние . Если активный синтез иммуноглобулинов в организме грудного ребёнка начинается после 4—6 мес. жизни, то такое состояние уже относят к патологическим и обозначают как поздний иммунный старт .
В норме антителообразование в организме ребёнка начинается после рождения, однако если после 20-й нед. беременности плод инфицируется вирусом краснухи, цитомегаловирусом, бледной трепонемой, токсоплазмами или другими микроорганизмами, то антителообразование начинается во внутриутробном периоде. Выявление повышенного уровня иммуноглобулинов у новорождённого, прежде всего IgM, указывает на пренатальную инфекцию (концентрацию IgM в крови пупочного канатика или у новорождённого 200 мг/л и более можно рассматривать как свидетельство антенатальной инфекции).
Первичные комбинированные иммунодефицитные состояния подразделяют на три группы: (1) тяжёлые комбинированные иммунодефициты , (2) комбинированные иммунодефициты с умеренновыраженным дефектом иммунного ответа и (3) минорные иммунодефицитные состояния .
Тяжёлые комбинированные иммунодефициты — иммунодефицитные состояния, при которых ребёнок погибает в первые месяцы или в первые годы жизни (такие дети редко живут более 1—2 лет). Единственная возможность терапии этих заболеваний — трансплантация костного мозга.
К этой группе относятся следующие болезни:
Ретикулярная дисгенезия проявляется аплазией кроветворной ткани. Блок дифференцировки при этом заболевании локализован уже на уровне стволовой кроветворной клетки. Дети погибают антенатально или вскоре после рождения от инфекционно-септических осложнений или злокачественных новообразований.
Синдром «голых» лимфоцитов — тяжёлый комбинированный иммунодефицит, при котором клетки организма, в том числе и лимфоциты не экспрессируют молекулы HLA-I . При этом становится невозможным Т-зависимый иммунный ответ. Количество Т- и В-лимфоцитов в крови нормальное. Заболевание манифестирует в возрасте 3—6 мес. в виде различных инфекций. Характерна задержка роста.
Болезнь Вискотта—Олдрича — иммунодефицит с тромбоцитопенией и экземой. Тип наследования — рецессивный, сцепленный с Х-хромосомой. Инфекционные процессы при этом заболевании развиваются, как правило, в конце первого года жизни. Результаты, полученные при изучении патогенеза синдрома Вискотта—Олдрича, ставят исследователей в тупик. В ранние сроки болезни органы иммунной системы не изменены, однако по мере её прогрессирования из тимуса и лимфоузлов корней лёгких (!) начинают исчезать лимфоциты. Наиболее выраженные изменения происходят в Т-системе иммунитета. Гуморальный ответ страдает меньше — снижается продукция IgM.
Синдром Гитлина — сочетание тяжёлого комбинированного иммунодефицита с недостаточностью продукции соматотропного гормона. Больные карликового роста. Заболевание также сопровождается незрелостью тимуса. Остановка его развития при синдроме Гитлина связана также с дефицитом гормона роста.
Болезнь Гланцмана—Риникера — тяжёлый иммунодефицит, описанный в 1950 г. швейцарскими врачами, именами которых названо заболевание. Смерть при отсутствии активной терапии наступает в большинстве случаев во второй половине первого года жизни, когда материнское молоко начинает вытесняться из рациона ребёнка другими продуктами. В первые же месяцы ребёнок получает с грудным молоком антитела, при этом он защищён пассивным иммунитетом. Масса тимуса уменьшена в 5—10 раз.
Синдром Гуда ( иммунодефицит с тимомой ) — первичный иммунодефицит, для которого характерна незрелость тимуса ( фетальный тимус ), в котором позже развивается опухоль из эпителиоцитов стромы ( тимома ). Изредка возникают злокачественные варианты этой опухоли. Характерна гипопластическая анемия .
Синдром Незелофа — первичный иммунодефицит, обусловленный гипо- или дисплазией тимуса. При этом в результате его функциональной недостаточности происходит нарушение дифференцировки Т-лимфоцитов.
Синдром Оменна описан в 1965 г. (G. S. Omenn) под названием семейный ретикулоэндотелиоз с эозинофилией . Он проявляется тяжёлым иммунодефицитом, поражением кожи по типу эритродермии и экземы, алопецией, хронической диареей, лимфаденопатией, гепатоспленомегалией, рецидивирующими респираторными инфекциями, лейкоцитозом (до 25 тыс. клеток в мкл) и эозинофилией крови. Характерна гипоплазия тимуса. Прогноз, как правило, неблагоприятный.
Патогенез синдрома связан с разрушением тканей и органов ребёнка пролиферирующими в его организме материнскими лимфоцитами. Обычно в кровь плода поступают единичные лимфоциты матери, но если таких клеток оказывается значительное количество и они составляют существенную массу лимфоидной ткани, то развивается реакция «трансплантат против хозяина» ( РТПХ ). В качестве трансплантата при этом синдроме выступают материнские лимфоциты. Особенно тяжёлые изменения развиваются в печени и в селезёнке, где под влиянием материнских лимфоцитов развиваются множественные мелкоочаговые некрозы . Синдром Оменна можно рассматривать как перинатальную форму РТПХ наряду со взрослой ( гомологичная болезнь ) и детской ( рант-болезнь ) формами.
Фермент аденозин-дезаминаза (АДА) у млекопитающих встречается во всех тканях, но наибольшая его концентрация выявляется в тимусе (в 10—15 раз больше, чем в других тканях). Поэтому дефицит или дефект этого энзима сопровождается прежде всего нарушениями в функционировании тимуса.
К комбинированным иммунодефицитам с умеренновыраженным дефектом иммунитета (при этих заболеваниях больные живут несколько десятилетий) относятся следующие синдромы:
Атаксия-телеангиэктазия Луи-Бар — наследственное заболевание, при котором, как правило, тяжёлого иммунодефицита не возникает, поэтому больные в среднем живут до 30—40 лет. Наиболее постоянный признак — низкий уровень или отсутствие IgA — встречается примерно у 70 % больных. Заболевание описано в 1941 году.
Помимо иммунодефицита развиваются следующие синдромы:
При наследственном цинк-зависимом иммунодефиците не происходит всасывания в тонкой кишке ионов цинка из-за дефекта специфического транспортного белка. Помимо комбинированного иммунодефицита развивается энтеропатический акродерматит с тяжёлым поражением кожи, алопецией, желудочно-кишечными расстройствами и неврологическими нарушениями, гипоплазией тимуса и плазматизацией ткани лимфоузлов. Введение цинка сульфата парентерально или перорально в больших дозах восстанавливает структуру вилочковой железы, ликвидирует вышеперечисленные признаки и предупреждает фатальный исход болезни. Цинк является кофактором многих ферментов, в том числе и такого распространённого в тканях организма как щёлочная фосфатаза . При дефиците цинка из органов иммунной системы страдает прежде всего тимус.
Для метафизарной хондродисплазии Мак-Кьюсика ( «гипоплазии хрящей и волос» ) характерны короткие конечности вследствие нарушения роста и созревания хрящевой ткани, тонкие скудные, лишённые пигмента волосы и умеренно выраженный (редко тяжёлый) комбинированный иммунодефицит. Особо тяжело протекает у этих больных ветряная оспа , хотя к другим вирусным инфекциям они относительно устойчивы. У части больных развивается гипопластическая анемия .
Основным заболеванием в этой группе является общая вариабельная иммунная недостаточность .
Общая вариабельная иммунная недостаточность (ОВИН) — иммунодефицит, при котором снижена продукция плазматическими клетками иммуноглобулинов различных классов, а также активность Т-звена иммунитета. Заболевание имеет наследственный характер, но проявляется через некоторое время после рождения, иногда на втором-третьем десятилетии жизни. Клинически ОВИН сопровождается хроническими и частыми острыми воспалительными процессами в ЛОР-органах и дыхательных путях.
Генетические дефекты, обусловливающие нарушение функции макрофагов (гистиоцитов) и микрофагов (нейтрофильных гранулоцитов), лежат в основе следующих заболеваний и синдромов:
I. Наследственные нейтропении
II. Наследственные дефекты хемотаксиса, фагоцитоза и бактерицидной активности клеток-скэвенджеров
Они проявляются рецидивирующими инфекциями, вызванными гноеродной микрофлорой (прежде всего, стафилококками и кишечными бактериями). Наиболее часто при этом поражаются кожа, лёгкие и слизистые оболочки полости рта и кишечника.
Среди наследственных нейтропений различают фатальный агранулоцитоз Костмана, циклическую и постоянную нейтропении. Под нейтропенией понимают уменьшение содержания нейтрофильных гранулоцитов в периферической крови менее 1500 клеток в мкл. Однако эта цифра, приемлемая для популяции в целом, тем не менее является условной, поскольку некоторые люди отличаются низким уровнем нейтрофилов без каких-либо патологических изменений в организме (это говорит о том, что обычно нейтрофильных гранулоцитов вырабатывается больше, чем требуется для эффективной антибактериальной защиты).
Агранулоцитоз Ко́стмана — тяжёлое заболевание, при котором миелоидная ткань не способна к продукции гранулоцитов, прежде всего нейтрофильных. Поскольку нейтрофильные гранулоциты являются центральным звеном антибактериальной защиты организма, при болезни Костмана возникающие бактериальные инфекции протекают тяжело и завершаются летальным исходом уже в первые месяцы жизни ребёнка.
Циклическая нейтропения — наследственное заболевание человека и серой шотландской овчарки, проявляющееся в циклических изменениях интенсивности гемопоэза. При этом выявляются периодические колебания уровня для всех форменных элементов крови, в том числе и для нейтрофильных гранулоцитов. Более того, для нейтрофилов с их долгим сроком созревания и коротким временем жизни характерны наиболее очевидные отклонения в виде 21-дневных подъёмов и снижений. В цикле моноцитов обратные взаимоотношения — количество этих клеток достигает пика во время снижения числа нейтрофилов. Увеличение и уменьшение концентрации эритроцитов и тромбоцитов едва заметны в связи со значительно большей продолжительностью их жизни.
Наследственная хроническая ациклическая нейтропения проявляется сразу после рождения, то есть является врождённой . Она наследуется в ряде случаев как аутосомно-доминантный признак, в других — как аутосомно-рецессивный . Может сопровождаться моноцитозом и эозинофилией (отдельно или в сочетании). От наследственных вариантов хронической ациклической нейтропении следует отличать приобретённые варианты , в частности аутоиммунную нейтропению (как проявление аутоиммунного агранулоцитоза или при других аутоиммунных болезнях), при циррозе печени вследствие гиперспленизма и транзиторную неонатальную нейтропению , развивающуюся при иммунной реакции матери на нейтрофильные гранулоциты плода. В некоторых случаях на фоне нейтропении возникают миелоидная лейкемия или апластическая анемия.
Ряд наследственных заболеваний характеризуются нарушением хемотаксиса, фагоцитоза и бактерицидной активности нейтрофильных гранулоцитов и макрофагов. Эти дефекты лежат в основе хронической гранулёматозной болезни детей, синдрома Миллера и ряда других заболеваний.
Хроническая гранулёматозная болезнь детей — наследственная недостаточность бактерицидной функции фагоцитов. Миграционная активность клеток и способность к фагоцитозу, как правило, не нарушены. Заболевание описано в 1957 г. Поглощённые клетками микроорганизмы не разрушаются в фаголизосомах, остаются жизнеспособными и активно размножаются ( эндоцитобиоз ). Дефект бактерицидной функции макрофагов и нейтрофилов обусловлен недостаточностью ферментов синтеза активных радикалов кислорода — НАДФ*Н-оксидаз , без участия которых фагоцит не способен разрушить микробную клетку. По той же причине (отсутствие или низкая концентрация кислородных метаболитов) формирующийся в повреждённой ткани гнойный экссудат не обладает литическими свойствами, поэтому для хронической гранулёматозной болезни детей не характерно разлитое гнойное воспаление ( флегмона ), а возникают абсцессы , чаще множественные микроабсцессы (пустулы и апостемы). В тканевых срезах, окрашенных гематоксилином и эозином, в цитоплазме макрофагов выявляется множество гранул золотистого пигмента ( цероида ). Пигментированные гистиоциты помогают поставить диагноз.
Синдром «ленивых лейкоцитов» Миллера — совокупность наследственных дефектов функции нейтрофильных гранулоцитов:
Синдром «ленивых лейкоцитов» в сочетании с врождённой недостаточностью экзокринной функции поджелудочной железы называется болезнью Шва́хмана (Шве́кмана), с полным альбинизмом — болезнью Чедьяка—Хигаси (при этом заболевании в цитоплазме нейтрофилов, макрофагов, моноцитов и лимфоцитов обнаруживаются гигантские азурофильные гранулы , а в меланоцитах происходит патологическая агрегация меланосом , лежащая в основе альбинизма).
Кроме того, описаны наследственные дефекты ферментов фагоцитов и селективные нарушения в функционировании элементов цитоскелета:
Первичная недостаточность миелопероксидазы нейтрофильных гранулоцитов и моноцитов/макрофагов наследуется по аутосомно-рецессивному типу. При этом не происходит синтеза из пероксида водорода других активных метаболитов кислорода (прежде всего гидроксильного радикала) и галоид-содержащих соединений. Эозинофильные гранулоциты не страдают. Клинические проявления болезни соответствуют хронической гранулёматозной болезни детей , но протекают значительно менее тяжело, так как фагоциты не теряют способность образовывать пероксид водорода . Кроме того, известны наследственные дефекты НАДН-оксидазы , глютатион-пероксидазы , глюкозо-6-фосфат-дегидрогеназы нейтрофильных гранулоцитов.
Первичный дефект полимеризации актина в нейтрофильных гранулоцитах характеризуется утратой ими способности к локомоции и фагоцитозу вследствие блокады процесса полимеризации актина, необходимого для образования псевдоподий и фагосом.
Наследственный дефект образования тафцина проявляется хроническими неспецифическими воспалительными заболеваниями лёгких и лимфоузлов. Синдром наследуется по аутосомно-доминантному типу. Тафцин — тетрапептид (тир—лиз—про—арг), высвобождающийся из молекулы IgG под воздействием специфических протеаз фагоцитов; он усиливает фагоцитарную активность нейтрофильных гранулоцитов.
Недостаточность белков комплемента проявляется по-разному в зависимости от того, какой (или какие) белки отсутствуют.
Выделяют три группы заболеваний, связанных с первичным дефицитом комплемента:
Комплемент-зависимые иммунодефицитные синдромы — заболевания, сопровождающиеся недостаточностью антибактериальной защиты организма. Они проявляются частыми инфекционными процессами в различных органах и тканях. Поскольку белки комплемента при активации играют роль хемоаттрактантов и опсонинов, обеспечивая эффективную функцию фагоцитирующих клеток, то при дефиците компонентов комплемента формируется вторичная недостаточность функции макрофагов и нейтрофильных гранулоцитов. Особенно часто инфекционные процессы при этом вызваны стрептококками, в частности пневмококками, и Haemophilus influenzae . В эту группу включают недостаточность С3b-инактиватора, белков С3, С6 и С8.
Недостаточность С3b-инактиватора. С3b-инактиватор играет роль ингибитора альтернативного пути активации комплемента. При его отсутствии происходит быстрое потребление С3-компонента ( вторичный дефицит С3 ), который в нормальных условиях принимает активное участие в антибактериальной защите организма. Белка С3 у больных в плазме примерно 20 % от нормы. Однако на 75 % он представлен С3b-фрагментом. Уровень нативного С3 составляет всего 5 % от нормы. Скорость расщепления С3 у больных повышена почти в 5 раз. Показано, что через 2 часа после инъекции нативного С3 расщеплению подвергается 40 % введённых молекул. Помимо вторичного дефицита С3 формируется вторичная недостаточность белка С5 , однако она менее выражена (примерно 40 % от нормального уровня). Заметно снижена концентрация фактора В — 5 % от нормы (расщепление фактора В происходит под влиянием фактора D ). Уровень пропердина снижен незначительно. Больные при этом заболевании страдают различными бактериальными инфекциями.
Недостаточность С3. Недостаточность С3-компонента комплемента также проявляется различными бактериозами. В основе заболевания, в отличие от недостаточности С3b-инактиватора, лежит первичный дефицит С3-белка.
Недостаточность белков комплемента провоцирует возникновение аутоиммунных заболеваний , прежде всего (1) красной волчанки , (2) так называемого волчаночно-подобного синдрома и (3) ревматоидного артрита . Часто поражаются почки по типу гломерулонефрита. У больных также описаны пурпура Шёнлейна—Геноха и полимиозит. К этим заболеваниям относятся недостаточность белков С1, С2, С4 и С5. Гены этих белков сцеплены с генами иммунного ответа ( генами МНС ), поэтому дефекты их, как правило, обоюдны.
Недостаточность С2. Недостаточность С2 является самым частым вариантом первичного дефицита белков комплемента. С2 синтезируют фиксированные и блуждающие макрофаги, фагоцитарная функция которых при этом не нарушена.
К третьей группе состояний, связанных с первичной недостаточностью комплемента, относится наследственный ангионевротический отёк Кви́нке—О́слера , в основе которого лежит недостаточность С1-ингибитора. У отдельных больных при этом возникают аутоиммунные процессы, прежде всего красная волчанка.