Interested Article - Синдром Смит — Магенис

- 2020-09-07
- 1
Синдром Смит — Магенис (СМС) — наследственное заболевание, имеет такие проявления, как умственная отсталость , аномалии лица, проблемы со сном и многочисленные поведенческие проблемы, в том числе и такие, как самоповреждение. Частота синдрома Смит — Магенис составляет 1 случай на 15 000-25 000 человек . Синдром Смит — Магенис, как правило, обусловлен микроделецией в коротком плече 17-ой хромосомы (17p), и иногда его называют 17p-синдром .
Симптомы
У маленького ребёнка с синдромом Смит — Магенис череп и мягкие ткани головы имеют следующие особенности: широкое и квадратное лицо, глубоко посаженные глаза, большие щеки и выступающую челюсть, а также плоская переносица. По мере взросления ребёнка переносица становится более выраженной, «трамплиновидной». Глаза, как правило, глубоко и близко друг к другу посажены. Брови широкие, густые, сросшиеся, расширяющиеся по бокам. Рот — самая заметная особенность, верхняя и нижняя губы полные, рот широкий. Рот изогнут вниз, а верхняя губа приподнята наружу, благодаря мясистому губному желобку . Эти черты лица становятся более заметными с возрастом, так как рост нижней челюсти опережает рост верхней, что приводит к явной гипоплазии средней части лица. Присутствует также лёгкая брахицефалия .
Для пациентов с синдромом Смит — Магенис с раннего возраста характерны нарушения сна. Больные могут быть очень сонными в течение дня, но испытывают проблемы с засыпанием и просыпаются несколько раз каждую ночь из-за инвертированного циркадного ритма выработки мелатонина .
Люди с синдромом Смит — Магенис очень общительны и, как правило, ласковы, но все они имеют множество поведенческих проблем. Поведенческие проблемы заключаются в частых вспышках гнева, агрессии, беспокойстве, компульсивном поведении, импульсивности и трудностях с обращением внимания. Самоповреждения, в том числе укусы, удары, удары по голове и ковыряние, очень распространены. Повторяющиеся самостоятельные обнимания — это поведенческая черта, которая может быть уникальной для синдрома Смит — Магенис. Люди с этим заболеванием могут также принудительно облизывать пальцы и перелистывать страницы книг и журналов (поведение, известное как «лизать и переворачивать»), а также обладать впечатляющей способностью вспомнить широкий спектр мелких деталей о людях или предметной области, пустяки.
Другие симптомы могут включать низкий рост, искривление позвоночника ( сколиоз ), хриплый голос, снижение чувствительности к боли и температуре. У некоторых людей с этим расстройством есть нарушения слуха, которые приводят к глухоте . Пострадавшие люди могут иметь нарушения зрения, которые вызывают близорукость , косоглазие и другие проблемы со зрением. У людей с синдромом Смит — Магенис также отмечались пороки сердца и почек, хотя они встречаются редко.
Генетика
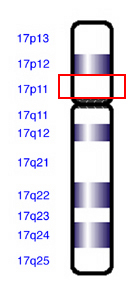
Синдром Смит — Магенис в большинстве случаев вызван хромосомной перестройкой , приводящей к утрате определённых сегментов одной хромосомой из пары 17 хромосом . У большинства людей с СМС происходит делеция генетического материала из определённой области 17 хромосомы (17p11.2). Хотя этот регион содержит несколько генов, недавно исследователи обнаружили, что потеря конкретного гена RAI1 отвечает за большинство характерных особенностей этого синдрома . Утрата соседних с RAI1 генов в хромосоме 17 усугубляет серьёзность клинических признаков и приводит к вариабельности проявлений синдрома. Потеря дополнительных генов в делетированном участке может помочь объяснить, почему особенности синдрома Смит — Магенис варьируют среди пораженных людей. У небольшой доли пациентов синдромом Смит — Магенис вызван мутацией в гене RAI1, а не делецией хромосомы 17.
Делеции хромосомы 17 и мутации гена RAI1 приводят к выработке аномальной или нефункциональной версии белка RAI1. RAI1 является транскрипционным фактором , который регулирует экспрессию множества генов, в том числе нескольких, которые участвуют в контроле циркадного ритма, таких как . Группы, возглавляемые (Медицинский колледж Бэйлора) и Сарой Элси (Университет Содружества Вирджинии), в настоящее время изучают роль этого гена в патогенезе синдрома Смит — Магенис .
СМС обычно не наследуются. Это состояние обычно возникает в результате мутации, происходящей во время образования репродуктивных клеток (яйцеклеток или сперматозоидов) или в начале эмбрионального развития. Люди с синдромом Смит — Магенис чаще всего не имеют истории заболевания в своей семье.
Диагностика
Синдром Смит — Магенис обычно подтверждается анализом клеток крови, который называют цитогенетическим анализом, или кариотипированием . Для диагностики используется метод, называемый FISH ( флуоресцентная гибридизация in situ ). Делеции небольшого размера (микроделеции) иногда оставались незамеченными в стандартном тесте FISH, что приводило к отрицательныму результату кариотипирования у пациентов с СМС. Недавняя разработка теста FISH на делецию в 17p11.2 позволила более точно обнаруживать это нарушение . Однако требуется дальнейшее разработка диагностики для выявления синдрома Смит-Магенис, вызванного мутацией гена RAI1 .
Детям с СМС часто ставят психиатрические диагнозы, такие как аутизм , синдром дефицита внимания и гиперактивности (СДВГ), обсессивно-компульсивное расстройство (ОКР), синдром дефицита внимания (СДВГ) и / или расстройства настроения .
Лечение
Лечение синдрома Смит — Магенис основано на управлении его симптомами. Дети с СМС часто нуждаются в нескольких формах поддержки, включая физиотерапию , трудотерапию и логопедию . Поддержка часто требуется в течение всей жизни больного.
Лекарства часто используются для устранения некоторых симптомов. Мелатониновые добавки и тразодон обычно используются для регуляции нарушений сна. В сочетании с экзогенным мелатонином блокада выработки эндогенного мелатонина в течение светлого времени суток адренергическим антагонистом может повысить концентрацию, улучшить время сна и время сна и помочь в улучшении поведения . Другие лекарства (такие как рисперидон ) иногда используются для регулирования агрессивного поведения.
Происхождение названия
Название «Смит — Магенис» происходит от фамилий двух женщин-учёных, которые в 1986 году описали это генетическое заболевание. Это Энн С. М. Смит, генетический консультант в Национальных институтах здравоохранения , и Рут Эллен Магенис , педиатр, медицинский генетик и цитогенетик из .
Примечания
- (декабрь 2013). Дата обращения: 27 марта 2016. 9 апреля 2016 года.
- Bi, W; Yan, J; Stankiewicz, P; Park, SS; Walz, K; Boerkoel, CF; Potocki, L; Shaffer, LG; Devriendt, K; Nowaczyk, MJ; Inoue, K; Lupski, J. R. Genes in a refined Smith-Magenis syndrome critical deletion interval on chromosome 17p11.2 and the syntenic region of the mouse. (англ.) // : journal. — 2002. — May ( vol. 12 , no. 5 ). — P. 713—728 . — doi : . — . — PMC .
- Allanson, Judith E.; Greenberg, Frank; Smith, Ann C. M. (англ.) // : journal. — 1999. — Vol. 36 . — P. 394—397 . 20 октября 2019 года.
- De Leersnyder H., De Blois M. C., Claustrat B., etal. Inversion of the circadian rhythm of melatonin in the Smith-Magenis syndrome (англ.) // : journal. — 2001. — Vol. 139 , no. 1 . — P. 111—116 . — doi : . — .
- Shaw, CJ; Withers, MA; Lupski, J. R. Uncommon deletions of the Smith-Magenis syndrome region can be recurrent when alternate low-copy repeats act as homologous recombination substrates. (англ.) // : journal. — 2004. — July ( vol. 75 , no. 1 ). — P. 75—81 . — doi : . — . — PMC .
- Girirajan S., Vlangos C. N., Szomju B. B., etal. Genotype-phenotype correlation in Smith–Magenis syndrome: evidence that multiple genes in 17p11.2 contribute to the clinical spectrum (англ.) // : journal. — 2006. — Vol. 8 , no. 7 . — P. 417—427 . — doi : . — .
- Elsea, SH; Girirajan, S. Smith-Magenis syndrome. (англ.) // : journal. — 2008. — April ( vol. 16 , no. 4 ). — P. 412—421 . — doi : . — .
- Williams S. R., Zies D., Mullegama S. V., etal. Smith-Magenis syndrome results in disruption of CLOCK gene transcription and reveals an integral role for RAI1 in the maintenance of circadian rhythmicity (англ.) // : journal. — 2012. — Vol. 90 , no. 6 . — P. 941—949 . — doi : . — . — PMC .
- . Дата обращения: 21 октября 2019. 28 июня 2021 года.
- . Дата обращения: 21 октября 2019. 21 октября 2019 года.
- Lupski, James R.; Potocki, Lorraine; Chen, Ken-Shiung; Park, Sung-Sup; Osterholm, Doreen E.; Withers, Marjorie A.; Kimonis, Virginia; Summers, Anne M.; Meschino, Wendy S.; Anyane-Yeboa, Kwame; Kashork, Catherine D.; Shaffer, Lisa G. Molecular mechanism for duplication 17p11.2— the homologous recombination reciprocal of the Smith-Magenis microdeletion (англ.) // Nature Genetics : journal. — 2000. — 1 January ( vol. 24 , no. 1 ). — P. 84—87 . — doi : . — .
-
De Leersnyder, H.
Inverted rhythm of melatonin secretion in Smith–Magenis syndrome: from symptoms to treatment
(англ.)
//
: journal. — 2006. — September (
vol. 17
,
no. 7
). —
P. 291—298
. —
doi
:
. —
.
De-Leersnyder H., de-Blois M. C., Vekemans M., Sidi D., Villain E., Kindermans C., Munnich A. (англ.) // : journal. — 2001. — September ( vol. 38 , no. 9 ). — P. 586—590 . — doi : . — . — PMC . 23 ноября 2006 года. - на Who Named It?
- Smith A. C., McGavran L., Robinson J., etal. Interstitial deletion of (17)(p11.2p11.2) in nine patients (англ.) // : journal. — 1986. — Vol. 24 , no. 3 . — P. 393—414 . — doi : . — .
Ссылки
| Аутосомные |
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| X / Y связанные |
|
||||||||
| Транслокации |
|
||||||||
| Иные | |||||||||
- 2020-09-07
- 1
