![{\displaystyle {\mathsf {R-COOH+ATP+CoA-SH{\xrightarrow[{}]{Mg^{2+}}}R-COS-CoA+AMP+H_{4}P_{2}O_{7}}}.}](/images/005/718/5718947/1.jpg?rand=844586)


![{\displaystyle {\left[({\frac {n}{2}}\cdot 10)+(({\frac {n}{2}}-1)\cdot 4)-2\right]}}](/images/005/718/5718947/10.jpg?rand=154018)
![{\displaystyle {\left[({\frac {n-3}{2}}\cdot 10)+(({\frac {n}{2}}-1,5)\cdot 4)+25-3\right]}}](/images/005/718/5718947/11.jpg?rand=869017)
Антиоксиданты
- 1 year ago
- 0
- 0

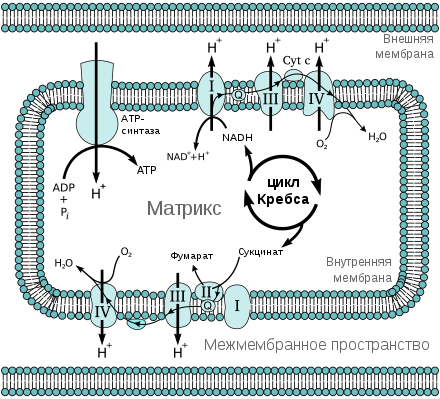
Бе́та-окисле́ние ( β-окисление ), также цикл Кноопа — Линена , — метаболический процесс . Своё название процесс получил по 2-му углеродному атому (С-3 или β-положение) от карбоксильной группы (-СООН) жирной кислоты , который подвергается окислению и последовательному отделению от молекулы. Продуктами каждого цикла β-окисления являются ФАД H 2 , НАДH и ацетил-КоА . Реакции β-окисления и последующего окисления ацетил-КоА в цикле Кребса служат одним из основных источников энергии для синтеза АТФ по механизму окислительного фосфорилирования .
В эукариотических клетках β-окисление происходит исключительно в аэробных условиях в матриксе митохондрий или пероксисомах , у растений этот процесс осуществляется в глиоксисомах .
Процесс β-окисления представляет собой специфический путь деградации жирных кислот. Он является одним из главных источников энергии, служащей для синтеза АТФ .
Все реакции многостадийного окисления ускоряются специфическими ферментами. β-Окисление высших жирных кислот является универсальным биохимическим процессом, протекающим во всех живых организмах. У млекопитающих этот процесс происходит во многих тканях, в первую очередь в печени , почках и сердце . Ненасыщенные высшие жирные кислоты (олеиновая, линолевая, линоленовая и др.) предварительно восстанавливаются до предельных кислот.
Помимо β-окисления, которое является основным процессом деградации жирных кислот у животных и человека, существуют ещё α-окисление и ω-окисление. α-Окисление встречается как у растений , так и у животных, однако, весь процесс происходит в пероксисомах . ω-Окисление менее распространено среди животных ( позвоночные ), встречается главным образом у растений . Процесс ω-окисления происходит в эндоплазматическом ретикулуме (ЭР).
β-Окисление было открыто в 1904 году немецким химиком ( Franz Knoop ) в опытах с кормлением собак различными жирными кислотами, в которых один атом водорода на концевом атоме ω-С углерода метильной группы -CH 3 был замещён на фенильный радикал -С 6 H 5 .
Францем Кноопом было выдвинуто предположение, что окисление молекулы жирной кислоты в тканях организма происходит в β-положении. В результате от молекулы жирной кислоты последовательно отщепляются двууглеродные фрагменты со стороны карбоксильной группы .
Жирные кислоты , входящие в состав естественных жиров животных и растений, имеют чётное число углеродных атомов. Любая такая кислота, от которой отщепляется по паре углеродных атомов, в конце концов проходит через стадию масляной кислоты . После очередного β-окисления масляная кислота становится ацетоуксусной . Последняя затем гидролизуется до двух молекул уксусной кислоты . Однако в то время механизмы окисления жирных кислот, происходящие при β-С атоме были ещё неизвестны . В 1948—1949 гг. Кеннеди и Ленинджер установили, что процесс окисления жирных кислот происходит в митохондриях . Ф. Линен с сотрудниками (1954—1958 гг.) описал основные ферментативные процессы окисления жирных кислот .
Теория β-окисления жирных кислот, предложенная Ф. Кноопом, в значительной мере послужила основой современных представлений о механизме окисления жирных кислот .
β-Окисление представляет собой последовательность процессов:
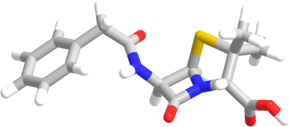
Жирные кислоты, которые образовались в клетке путём гидролиза триацилглицеридов или поступившие в неё из крови должны быть активированы, так как сами по себе они являются метаболическими инертными веществами, и вследствие этого не могут быть подвержены биохимическим реакциям, включая и окисление. Процесс их активирования происходит в цитоплазме при участии АТФ , кофермента A (HS-СoA) и ионов Mg 2+ . Реакция катализируется ферментом ацил-КоА-синтетазой жирных кислот с длинной цепью ( Long-chain-fatty-acid—CоА ligase , КФ ), процесс является эндергоническим , то есть протекает за счёт использования энергии гидролиза молекулы АТФ :
ацил-КоА-синтетазы находятся как в цитоплазме , так и в матриксе митохондрий. Эти ферменты отличаются по специфичности к жирным кислотам с различной длиной углеводородной цепи. Жирные кислоты с короткой и средней длиной цепи (от 4 до 12 атомов углерода) могут проникать в матрикс митохондрий путём диффузии . Активация этих жирных кислот происходит в матриксе митохондрий .
Жирные кислоты с длинной цепью, которые преобладают в организме человека (от 12 до 20 атомов углерода), активируются ацил-КоА-синтетазами, расположенными на внешней стороне внешней мембраны митохондрий.
Выделившийся в ходе реакции пирофосфат гидролизуется ферментом пирофосфатазой ( КФ ):
При этом происходит сдвиг равновесия реакции в сторону образования ацил-КоА .
Поскольку процесс активации жирных кислот происходит в цитоплазме, то далее необходим транспорт ацил-КоА через мембрану внутрь митохондрии.
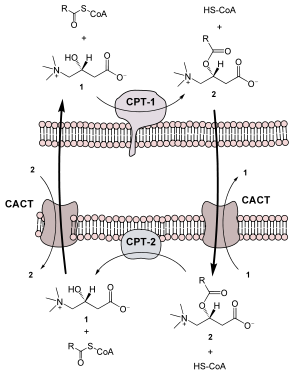
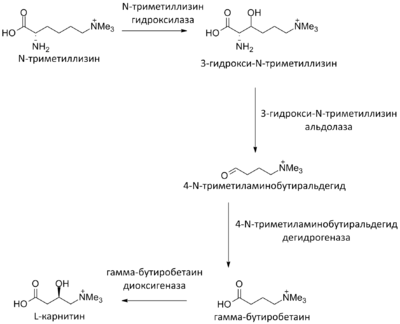
Транспортировка жирных кислот с длинной цепью через плотную митохондриальную мембрану осуществляется посредством карнитина . В наружной мембране митохондрий находится фермент карнитинацилтрансфераза I ( карнитин-пальмитоилтрансфераза I , CPT1, КФ ), катализирующий реакцию с образованием ацилкарнитина (ацильная группа переносится с атома серы КоА на гидроксильную группу карнитина с образованием ацилкарнитина (карнитин-СOR)), который диффундирует через наружную митохондриальную мембрану :
R-CO~SКоА + карнитин ↔ карнитин-COR + КоА-SH
Образовавшийся ацилкарнитин проходит через межмембранное пространство к наружной стороне внутренней мембраны и транспортируется с помощью фермента карнитин-ацилкарнитин-транслоказы (CACT) .
После прохождения ацилкарнитина (карнитин-СOR) через мембрану митохондрии происходит обратная реакция — расщепление ацилкарнитина при участии КоА-SH и фермента митохондриальной карнитинацил-КоА-трансферазы или карнитинацилтрансферазы II ( карнитин-пальмитоилтрансфераза II , CPT2, КФ ):
КоА-SH + карнитин-COR ↔ R-CO~SКоА + карнитин
Таким образом, ацил-КоА становится доступным для ферментов β-окисления. Свободный карнитин возвращается на цитоплазматическую сторону внутренней мембраны митохондрии той же транслоказой .
После этого ацил-КоА включается в реакции β-окисления.
Процесс трансмембранного переноса жирных кислот может ингибироваться малонил-КоА .
В матриксе митохондрии происходит окисление жирных кислот в цикле Кнооппа — Линена. В нём участвуют четыре фермента, которые последовательно действуют на ацил-КоА. Конечным метаболитом данного цикла является ацетил-КоА . Сам процесс состоит из четырёх реакций.
| Наименование реакции | Схема реакции | Фермент | образовавшийся продукт |
|---|---|---|---|
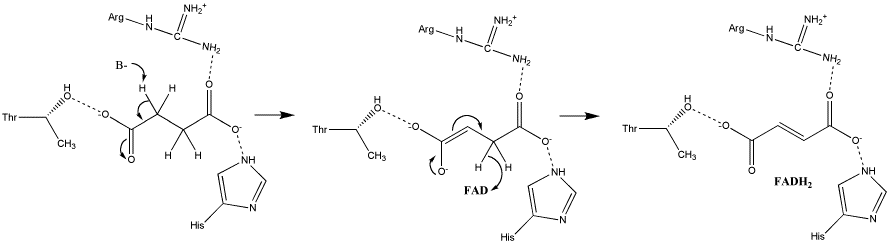
| Дегидрирование активированной жирной кислоты (ацил-КоА) . β-Окисление начинается с дегидрирования ацил-КоА ФАД-зависимой ацил-КоА дегидрогеназой жирных кислот с длинной цепью (LCAD) с образованием двойной связи между α- и β-атомами углерода (С-2 и С-3) в продукте реакции — еноил-КоА. Восстановленный в этой реакции кофермент ФАДH 2 передаёт атомы водорода в ЭТЦ на кофермент Q . В результате синтезируются 2 молекулы ATФ . |

|
ацил-КоА-дегидрогеназа ( КФ ) | Транс-Δ 2 -еноил-КоА |
| Реакция гидратации . Ненасыщенный ацил-КоА (еноил-CоА) при участии фермента еноил-CоА-гидратазы присоединяет молекулу воды . В результате образуется β-гидроксиацил-КоА. Реакция обратима и стереоспецифична, образовавшийся продукт имеет L-форму. |

|
Еноил-CоА-гидратаза (КФ ) | L-β-гидроксиацил-КоА |
| НАД + — зависимое окисление или вторая реакция дегидрирования . Образовавшийся L-β-гидроксиацил-КоА затем окисляется. Реакция катализируется НАД + -зависимой дегидрогеназой. |

|
L-β-гидроксиацетилдегидрогеназа (КФ ) | L-β-кетоацил-КоА |
| Тиолазная реакция . В этой реакции β-кетоацил-КоА взаимодействует с коферментом А . В результате происходит расщепление β-кетоацил-КоА и образуется укороченный на два углеродных атома ацил-КоА и двууглеродный фрагмент в виде ацетил-КоА. Данная реакция катализируется ацетил-КоА-ацилтрансферазой (или β-кетотиолазой). |
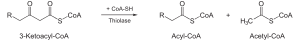
|
β-Кетотиолаза (КФ ) | Ацил-КоА и ацетил-КоА |
Образовавшийся ацетил-КоА подвергается окислению в цикле Кребса, а ацил-КоА, укоротившийся на два углеродных атома, снова многократно проходит весь путь β-окисления вплоть до образования бутирил-КоА (4-углеродное соединение), который в свою очередь окисляется до 2 молекул ацетил-КоА. ФАДH 2 и НАДH·H поступают прямо в дыхательную цепь .
Для полной деградации длинноцепочечной жирной кислоты цикл должен многократно повторяться, так, например, для стеарил-CоА (С 17 Н 35 СО~SКоА) необходимы восемь циклов .
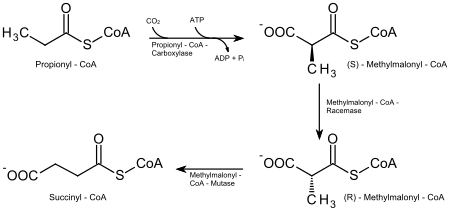
В результате окисления жирных кислот с нечётным числом углеродных атомов образуются не только ацетил-КоА, ФАД H 2 и НАДH , но и одна молекула пропионил-КоА (C 2 H 5 -CO~SКоА).
Пропионил-КоА превращается в сукцинил-КоА последовательно. Карбоксилирование пропионил-КоА осуществляется под действием пропионил-КоA-карбоксилазы ( КФ ) (коферментом этого фермента служит биотин (витамин B7) — переносчик карбоксигрупп; реакция требует также АТФ ). Реакция стереоспецифична. Продуктом реакции является S-изомер метилмалонил-КоА, который катализируется метилмалонил-КоА-рацемазой ( КФ ) в R-изомер. Образовавшийся R-изомер метилмалонил-КоА под действием фермента метилмалонил-КоА-мутазы ( КФ ) (кофермент которой дезоксиаденозилцианокобаламин является производным витамина B12 ) превращается в сукцинил-КоА, который далее вступает в цикл Кребса .
Ненасыщенные жирные кислоты (НЖК) составляют почти половину от общего количества жирных кислот в организме человека. Особенности β-окисления таких кислот определяются положением и числом двойных связей. Двойные связи (-C=C-) природных ненасыщенных жирных кислот ( олеиновой , линолевой и т. д.) имеют цис-конфигурацию, а в КоА-эфирах ненасыщенных кислот, являющихся промежуточными продуктами при β-окислении насыщенных жирных кислот, двойные связи имеют транс-конфигурацию. β-Окисление этих кислот идёт обычным путём до тех пор, пока двойная связь не окажется между третьим и четвёртым атомами углерода. Затем фермент еноил-КоА-изомераза ( КФ ) перемещает двойную связь из положения Δ 3-4 в положение Δ 2-3 и изменяет цис-конформацию двойной связи на транс-, которая требуется для β-окисления. Далее процесс протекает также, как и для насыщенных кислот .
При окислении жирных кислот, имеющих две (-С=C-C-C=C-) и более ненасыщенные связи, требуется ещё один дополнительный фермент β-гидроксиацил-КоА-эпимераза ( КФ ).
Скорость окисления ненасыщенных жирных кислот много выше, чем насыщенных, что обусловлено наличием двойных связей. Например, если взять за эталон скорость окисления насыщенной стеариновой кислоты , то скорость окисления олеиновой в 11, линолевой в 114, линоленовой в 170, а арахидоновой почти в 200 раз выше, чем стеариновой .
В результате переноса электронов по ЭТЦ от ФАД H 2 и НАДH синтезируется по 4 молекулы АТФ (1,5 от ФАДH 2 , и 2,5 от НАДH ). В случае окисления пальмитиновой кислоты проходит 7 циклов β-окисления (16/2-1 = 7), что ведёт к образованию 4×7 = 28 молекул АТФ. В процессе β-окисления пальмитиновой кислоты образуется 8 молекул ацетил-КоА, каждая из которых при полном сгорании в цикле трикарбоновых кислот даёт 10 молекул АТФ, а 8 молекул дадут 10×8 = 80 молекул АТФ.
Таким образом, всего при полном окислении пальмитиновой кислоты образуется 28+80 = 108 молекул АТФ. Однако с учётом одной молекулы АТФ , которая гидролизуется до АМФ , то есть тратятся 2 макроэргические связи или две АТФ, в самом начале на процесс активирования (образования пальмитоил-CоА), общий энергетический выход при полном окислении одной молекулы пальмитиновой кислоты в условиях животного организма составит 108-2=106 молекул .
Суммарное уравнение окисления пальмитиновой кислоты выглядит следующим образом:
Формула для расчёта общего количества АТФ, которые генерируются в результате процесса β-окисления жирных кислот с чётным числом углеродных атомов:
где n — количество атомов углерода в молекуле жирной кислоты; 10 и 4 — соответственно, количество молекул АТФ, синтезируемых при окислении 1 моль ацетил-КоА в цикле Кребса и в одном цикле бета-окисления жирной кислоты (1,5 АТФ от ФАДН 2 и 2,5 АТФ от НАДН); -2 — количество АТФ, затраченные на активацию жирной кислоты.
Формула для расчёта общего количества АТФ, которые генерируются в результате процесса β-окисления жирных кислот с нечётным числом углеродных атомов:
где n — количество атомов углерода в молекуле жирной кислоты; 25 — количество АТФ, которое синтезируется при окислении образовавшейся 1 молекулы сукцинил-КоА в цикле Кребса до оксалоацетата С 4 (5 моль) и его последующего полного окисления в ЦТК (2×10 = 20 моль); -3 — количество АТФ, затраченные на активацию жирной кислоты (2 моль), а также на синтез метилмалонил-КоА (1 моль). Рассчитаем количество синтезируемых молекул АТФ при окислении 1 моль нонадекановой кислоты (С 19 ). Воспользуемся формулой: (19-3/2 × 10) + (19/2-1,5 × 4) + 25 - 3 = 134 моль АТФ.
Энергетический расчёт β-окисления для некоторых жирных кислот представлен в виде таблицы.
| Жирная кислота | Кол-во молекул АТФ генерируемых на 1 молекулу жирной кислоты | Кол-во затраченных молекул АТФ | Общий энергетический выход молекул АТФ |
|---|---|---|---|
| Каприловая кислота C 7 H 15 COOH | 44 | 2 | 44-2=42 |
| Лауриновая кислота С 11 Н 23 COOH | 80 | 2 | 80-2=78 |
| Миристиновая кислота С 13 Н 27 СООН | 94 | 2 | 94-2=92 |
| Пентадециловая кислота С 14 Н 29 СООН | 101 | 2 | 101-2=99 |
| Пальмитиновая кислота С 15 Н 31 СООН | 108 | 2 | 108-2=106 |
| Маргариновая кислота С 16 Н 33 СООН | 115 | 2 | 115-2=113 |
| Стеариновая кислота С 17 Н 35 СООН | 122 | 2 | 122-2=120 |
| Арахиновая кислота С 19 Н 39 СООН | 136 | 2 | 136-2=134 |
Во многих тканях окисление жирных кислот — важный источник энергии. Это ткани с высокой активностью ферментов цикла Кребса и цепи переноса электронов — клетки красных скелетных мышц , сердечная мышца (миокард), почки . Например, эритроциты , в которых отсутствуют митохондрии, не могут окислять жирные кислоты. В то же время жирные кислоты не могут служить источником энергии для мозга и других нервных тканей , так как они не проходят через гематоэнцефалический барьер , вследствие их гидрофобных свойств. Скорость обмена жирных кислот в нервных тканях на порядок ниже чем, например, в скелетных мышцах. Поэтому в таких ситуациях, особенно при длительном голодании, печень перерабатывает около 50 % поступающих в неё жирных кислот в другие источники энергии — кетоновые тела , которые может утилизировать нервная ткань .
Помимо β-окисления жирных кислот, происходящего в митохондриях существует и внемитохондриальное окисление. Жирные кислоты, имеющие бóльшую длину цепи (от С 20 ), не могут быть окислены в митохондриях из-за наличия плотной двойной мембраны, которая воспрепятствует процессу переноса их через межмембранное пространство. Поэтому окисление длиноцепочечных жирных кислот (С 20 -С 22 и более) происходит в пероксисомах . В пероксисомах процесс β-окисления жирных кислот протекает в модифицированном виде. Продуктами окисления в данном случае являются ацетил-КоА, октаноил-КоА и пероксид водорода Н 2 О 2 . Ацетил-КоА образуется на стадии, катализируемой ФАД-зависимой дегидрогеназой. Ферменты пероксисом не атакуют жирные кислоты с короткими цепями, и процесс β-окисления останавливается при образовании октаноил-КоА.
Данный процесс не сопряжён с окислительным фосфорилированием и генерацией АТФ и поэтому октаноил-КоА и ацетил-КоА переходят с КоА на карнитин и направляются в митохондрии, где окисляются с образованием АТФ .
Активация пероксисомального β-окисления происходит при избыточном содержании в потребляемой пищи жирных кислот начиная с С 20 , а также при приёме гиполипидемических лекарственных препаратов.
Скорость регуляции процесса β-окисления включает несколько факторов:
Скорость β-окисления зависит также от активности фермента карнитин-пальмитоилтрансферазы I (CPTI). В печени этот фермент ингибируется малонил-КоА, веществом, образующимся при биосинтезе жирных кислот .
В мышцах карнитин-пальмитоилтрансфераза I (CPTI) также ингибируется малонил-КоА. Хотя мышечная ткань не синтезирует жирные кислоты, в ней имеется изофермент ацетил-КоА-карбоксилазы, синтезирующий малонил-КоА для регуляции β-окисления. Данный изофермент фосфорилируется протеинкиназой А , которая активируется в клетках под действием адреналина , и АМФ-зависимой протеинкиназой и таким образом происходит его ингибирование; концентрация малонил-КоА снижается. Вследствие этого, при физической работе, когда в клетке появляется АМФ , под действием адреналина активируется β-окисление, однако, его скорость зависит ещё и от доступности кислорода. Поэтому β-окисление становится источником энергии для мышц только через 10-20 минут после начала физической нагрузки (так называемые аэробные нагрузки), когда приток кислорода к тканям увеличивается .
Дефекты карнитиновой транспортной системы проявляются в ферментопатиях и дефицитных состояний карнитина в организме человека.
Дефицитные состояния карнитина
Наиболее распространены дефицитные состояния, связанные с потерей карнитина во время некоторых состояний организма:
Признаками и симптомами недостатка карнитина являются приступы гипогликемии, возникающие из-за снижения глюконеогенеза в результате нарушения процесса β-окисления жирных кислот, уменьшение образования кетоновых тел, сопровождающееся повышением содержания свободных жирных кислот (СЖК) в плазме крови, мышечная слабость (миастения), а также накопление липидов .
При дефекте гена карнитин-пальмитоилтрансферазы I — СРТ1 (гораздо реже гена СРТ2) развивается печёночная форма недостаточности фермента, которая приводит к гипогликемии и понижению содержания кетоновых тел в плазме крови . Дефект гена карнитин-пальмитоилтрансферазы II СРТ2 вызывает у взрослых миопатии (периодические мышечные боли , мышечная слабость, подёргивания, миоглобинурия ), у новорождённых — фатальную печёночную форму (гипераммониемия, увеличенная активность сывороточных трансаминаз, гепатомегалия, некетотическая гипогликемия, кома ). Для недостаточности карнитин-пальмитоилтрансферазы II также характерна кардиомегалия .
Генетические нарушения ацил-КоА-дегидрогеназ жирных кислот средней цепи
В митохондриях имеется 3 вида ацил-КоА-дегидрогеназ , окисляющих жирные кислоты с длинной, средней или короткой цепью радикала. Жирные кислоты по мере укорочения радикала в процессе β-окисления могут последовательно окисляться этими ферментами. Генетический дефект дегидрогеназы жирных кислот со средней длиной радикала ( КФ ) — MCADD (сокр. от М edium- c hain a cyl-СоА d ehydrogenase d eficiency) наиболее распространён по сравнению с другими наследственными заболеваниями — 1:15 000. Частота дефектного гена ACADM , кодирующего ацил-КоА-дегидрогеназы жирных кислот со средней длиной цепи, среди европейской популяции — 1:40. Это аутосомно-рецессивное заболевание , возникающее в результате замены нуклеотида Т ( тимин ) на А ( аденин ) в 985-й позиции гена . Проявляется в накоплении жирных кислот средней цепи (особенно каприловой ) и их производных в крови и вторичным дефицитом карнитина. Характерными симптомами являются приступы рвоты , летаргическое состояние , сильнейшая некетотическая гипогликемия, вызванная обильной утилизацией глюкозы (особенно опасна для новорожденных), может развиться кома и возможен летальный исход. Большую опасность болезнь представляет у детей, так как среди них наблюдается самая большая летальность (до 60 %) .
Генетические нарушения ацил-КоА-дегидрогеназ жирных кислот с очень длинной углеродной цепью
Аутосомно-рецессивное тяжёлое генетическое заболевание встречается с частотой 1:3000-1:50000 у новорожденных стран Европы и США. Обусловлено мутацией гена ACADVL , который кодирует ацил-КоА-дегидрогеназу жирных кислот с очень длинной углеродной цепью — VLCAD (сокр. от V ery l ong c hain a cyl-CоА d ehydrogenase, КФ ). Данный фермент участвует в митохондриальном β-окислении жирных кислот, углеродная цепь которых содержит 14—20 атомов. Болезнь характеризуется накоплением жирных кислот (С 14 -С 20 ) в организме. Негативные проявления выражаются в поражениях тканей головного мозга ( энцефалопатии ), сердца ( кардиомиопатии ), печени ( жировая инфильтрация ). Симптомы схожи с MCADD. Существуют несколько форм дефицита ацил-КоА-дегидрогеназы жирных кислот с очень длинной углеродной цепью:
Системная форма встречается часто у новорожденных или детей раннего возраста и имеет самую высокую летальность (до 30 %). Наиболее тяжёлая и опасная форма заболевания.
Печёночная форма также часто обладает ранней манифестацией (развитием клинических проявлений), однако, имеет менее тяжёлое течении и летальность. Характеризуется приступами гипокетотической гипогликемии.
Миопатическая форма наблюдаются у детей школьного возраста и взрослых. Её основные проявления: непереносимость физической нагрузки ( миастения ), боли в мышцах (миалгии, рабдомиалгии), рабдомиолиз, изменение цвета мочи вследствие миоглобинурии .
Дикарбоновая ацидурия заболевание, связанное с повышенной экскрецией С 6 -С 10 -дикарбоновых кислот и возникающей на этом фоне гипогликемии , однако, не связанная с повышением содержания кетоновых тел. Причиной данного заболевания является MCADD. При этом нарушается β-окисление и усиливается ω-окисление длинноцепочечных жирных кислот, которые укорачиваются до среднецепочечных дикарбоновых кислот , выводимых из организма .
Синдром Целлвегера (Зеллвегера) или цереброгепаторенальный синдром, редкое наследственное заболевание описано американским педиатром Гансом Целлвегером ( англ. H.U. Zellweger ), которое проявляется в отсутствии пероксисом во всех тканях организма. Вследствие этого в организме, особенно в мозгу накапливаются полиеновые кислоты (С 26 —С 38 ), представляющие собой длиноцепочечные жирные кислоты . Примерная заболеваемость нарушениями биогенеза пероксисом спектра синдрома Целлвегера составляет 1:50 000 новорождённых в США и 1:500 000 новорождённых в Японии. Для синдрома характерны: пренатальная задержка роста; мышечная гипотония; затруднение сосания; арефлексия; долихоцефалия; высокий лоб; круглое плоское лицо; одутловатые веки; гипертелоризм; монголоидный разрез глаз; катаракта ; пигментная ретинопатия или дисплазия зрительного нерва; колобома радужки; низко расположенные ушные раковины; микрогнатия ; расщелина неба; латеральное или медиальное искривление пальцев; поражение печени ( гепатомегалия (увеличение объёма печени), дисгинезия внутрипеченочных протоков, цирроз печени ); поликистоз почек; нередко — тяжёлые, несовместимые с жизнью аномалии лёгких и пороки сердца; задержка психомоторного развития; судороги ; стойкая желтуха. При патоморфологическом исследовании выявляют задержку миелинизации нейронов; накопление липидов в астроцитах; в печени, почках и мозге уменьшено содержание плазмогенов; в клетках печени и других тканях организма снижено количество пероксисом, большинство пероксисомных ферментов неактивны. В крови повышена активность трансаминаз и отмечается стойкая гипербилирубинемия . Нарушения биогенеза пероксисом обусловлены мутациями в одном из 12 генов PEX , кодирующих пероксины. Мутации в этих генах ведут к аномалиям биогенеза пероксисом. Все варианты синдрома Целлвегера наследуются по аутосомно-рецессивному типу .
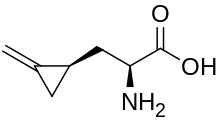
Специфическая болезнь, характеризуется сильнейшей интоксикацией, сопровождающаяся рвотой , гиповолемическим шоком, конвульсиями , гипогликемией , в тяжёлой форме может наступить кома и смертельный исход. Вызывается при употреблении:
В результате метаболизма гипоглицин превращается в метиленциклопропанацетил-КоА (мцпа-КоА), который инактивирует ацил-КоА-дегидрогеназу, вследствие чего ингибируется процесс β-окисления . Помимо этого мцпа-КоА способен блокировать ферменты глюконеогенеза . В присутствии гипоглицина происходит накопление главным образом бутирил-КоА, который гидролизуется до свободной масляной кислоты (бутирата). Масляная кислота в избытке попадает в кровь , косвенно вызывая гипогликемию .
|
|
|
|---|