Interested Article - Недостаточность кислой фосфатазы

- 2020-05-19
- 1
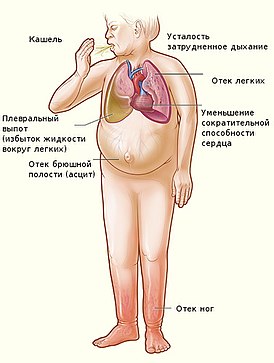
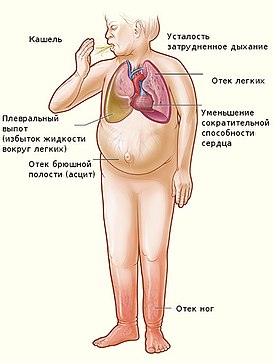
Недоста́точность ки́слой фосфата́зы — редкое наследственное заболевание из группы лизосомных болезней накопления . Клиническая симптоматика дефицита кислой фосфатазы развивается в раннем возрасте (до трёх лет жизни). Характеризуется гепатомегалией и задержкой психического развития .
Историческая справка
О первых пациентах сообщил Henry L. Nadler с соавторами в 1970 году: была выявлена недостаточность кислой фосфатазы лизосом в ФГА-стимулированных лимфоцитах и культивируемых фибробластах кожи . Уровень активности фермента составлял 20% от нормы в цельном гомогенате фибробластов и примерно 1% от нормы в лизосомальной фракции .
Пренатальная диагностика осуществлялась методом исследования уровня активности кислой фосфатазы в культивируемых клетках амниотической жидкости . Сообщалось о втором синдроме, протекающем на фоне низкого уровня кислой фосфатазы и клинически проявлявшемся опистотонусом и склонностью к кровотечению .
Наследование

Недостаточность кислой фосфатазы наследуется, как и подавляющее большинство лизосомных болезней накопления , по аутосомно-рецессивному типу наследования . Следовательно, с одинаковой частотой встречается как у мужчин , так и у женщин . Заболевание клинически манифестирует только в случае, когда обе аутосомы , полученные по одной от отца и матери, являются дефектными по гену ACP2 (повреждение обеих копий гена, находящихся на гомологичных аутосомах 11p11.2 ).
Патогенез
Развитие клинической симптоматики обусловлено генетическим дефектом — мутацией гена ACP2, расположенного на коротком плече 11-й хромосомы ( локус 11p11.2), кодирующего кислую фосфатазу .
Клиническая картина
Клиническая картина, описанная у нескольких пациентов , характеризовалась периодической рвотой , гипотензией , летаргией , опистотонусом , терминальным кровотечением и летальным исходом на 1-м году жизни . На фоне выявляемой гепатомегалии , в результате биопсии печени определяются широко распространённые очаги некроза клеток печени: гепатоциты увеличены в размере, цитоплазма вакуолизирована .
Прогноз
Неблагоприятный. Летальный исход наступает на первом году жизни .
См. также
Примечания
- Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- ↑ . Глава 316. Лизосомные болезни накопления (с. 250—273) . med-books.info. Дата обращения: 17 января 2015. 7 июня 2015 года.
- ↑ (англ.) . Deficiency of lysosomal acid phosphatase: a new familial metabolic disorder. New Eng. J. Med. 282: 302-307, 1970. PubMed: 5410815 . nejm.org. Дата обращения: 17 января 2015. 17 марта 2016 года.
- Nadler, H. L. Treatment of acid phosphatase deficiency disorders. Birth Defects Orig. Art. Ser. 9(2): 195-197, 1973. PubMed: 4139985 (англ.)
- ↑ Nadler, H. L. Acid phosphatase deficiency.In: Hers, H. G.; Van Hoof, F. : Lysosomes and Storage Disease. New York: Academic Press (pub.) 1972. (англ.)
- Nadler, H. L. Personal Communication. Chicago, Ill. 2/1978. (англ.)
- Nadler, H. L. Personal Communication. Detroit, Mich. 4/5/1982. (англ.)
- ↑ . Нарушения обмена липидов: недостаточность кислой фосфатазы . medkurs.ru. Дата обращения: 17 января 2015. 4 марта 2016 года.
- OMIM (англ.)
- OMIM (англ.)
Ссылки
- 2020-05-19
- 1