Атрофия
- 1 year ago
- 0
- 0

Спина́льная мы́шечная атрофи́я ( СМА ; англ. spinal muscular atrophy , SMA ) — разнородная группа наследственных заболеваний , протекающих с поражением / потерей двигательных нейронов передних рогов спинного мозга .
В большинстве случаев СМА вызвана мутациями в генах SMN1 и SMN2, кодирующих белок SMN, участвующий в синтезе сплайсосомы , и имеет аутосомно-рецессивный тип наследования . Реже встречаются варианты СМА, которые вызваны мутациями других генов; некоторые из этих вариантов имеют аутосомно-доминантный или X-сцепленный тип наследования .
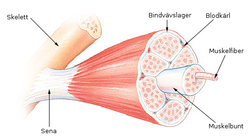
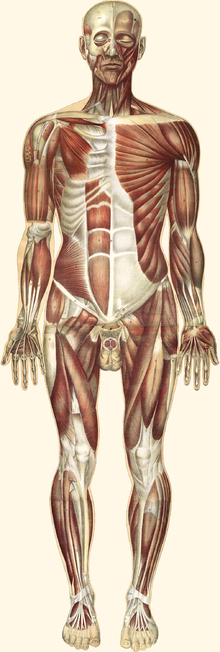
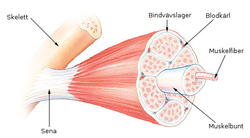
Для спинальных (их причина находится в спинном мозге) мышечных атрофий характерно нарушение работы поперечнополосатой мускулатуры ног, а также головы и шеи. У больных отмечаются нарушения произвольных движений — ползания младенцев, ходьбы, удержания головы, глотания. Мышцы рук обычно не страдают. Для спинальных характерно сохранение чувствительности, а также отсутствие задержки психического развития.
Спинальная мышечная атрофия у детей впервые была описана в 1891 году ( G. Werdnig ). Он представил описание патоморфологических изменений различных групп мышц, периферических нервов и спинного мозга, отметив симметричную атрофию клеток передних рогов спинного мозга и передних корешков. В 1892 г. обосновал нозологическую самостоятельность заболевания. В дальнейшем Вердниг и Хофман (1893) доказали, что заболевание сопровождается дегенерацией клеток передних рогов спинного мозга. В 1956 г. Э. Кугельберг и выделили новую спинальной мышечной атрофии, которая характеризуется более поздним началом и относительно доброкачественным течением по сравнению с описанной Верднигом и Хофманом.
СМА обычно вызвана мутацией в гене , который в норме производит белок SMN. Из-за мутации гена у людей с СМА производится меньшее количество белка SMN, что приводит к потере моторных нейронов.
Выделяют четыре формы проксимальной спинальной амиотрофии на основе возраста начала, тяжести течения и продолжительности жизни .
| Тип | Эпоним | Обычный возраст начала | Описание | OMIM |
|---|---|---|---|---|
| I: Младенческий | СМА I, болезнь Верднига-Хоффмана | 0-6 мес. | Наиболее неблагоприятная форма СМА. Дети испытывают недостаток моторного развития, имеют трудности с дыханием, затруднения с сосанием и глотанием, не держат голову, не сидят самостоятельно. | |
| II: Промежуточный | СМА II, болезнь Дубовица | 7-18 мес. | Больные этой формой спинальной амиотрофии дети могут есть, сидеть, но никогда не достигают способности ходить самостоятельно. Прогноз в этих случаях зависит от степени вовлечения в патологический процесс респираторных мышц. | |
| III: Юношеский | СМА III, болезнь Кюгельберга-Веландер | >18 мес. | Наименее опасная форма СМА детского возраста. Пациент способен стоять, но испытывает сильную слабость, с тенденцией к инвалидизации (передвижение в коляске). | |
| IV: Взрослый | СМА IV или взрослая форма | после 35 лет | Значительно не влияет на продолжительность жизни. Слабость проксимальной мускулатуры, фасцикуляции, снижение сухожильных рефлексов. Больные не способны ходить самостоятельно. |
Радикального лечения не существует.
Так как спинальная мышечная атрофия — нарушение, которое проявляется в синапсах мотонейронов , состояние может быть улучшено за счёт увеличения уровня белка SMN. Цель современных исследований — поиск препаратов, увеличивающих уровни SMN. Основные результаты получены [ источник не указан 3451 день ] пока в исследовательских группах США , Германии , Италии .
Предложено несколько препаратов ( вальпроевая кислота , и др.), проводятся их клинические исследования в группах добровольцев. Сведений о результативном применении стволовых клеток пока нет.
Больные СМА нуждаются в специальном диетическом питании, поддерживающей терапии и многих других попечительских действиях, иначе отмечается усиление отрицательной динамики.
В декабре 2016 в США был одобрен первый специализированный препарат для лечения СМА любого типа — «Спинраза» (Spinraza, нусинерсен). Нусинерсен (nusinersen), разработанный «Байоджен» (Biogen) и «Айонис фармасьютикал» (Ionis Pharmaceuticals), представляет собой антисмысловой олигонуклеотид для альтернативного сплайсинга пре- мРНК гена SMN2, который почти идентичен SMN1, и потому синтез полноразмерного белка SMN усиливается. «Спинраза» вводится интратекально, то есть в спинномозговой канал .
В мае 2019 года FDA США одобрило «Золгенсма» ( Zolgensma , онасемноген абепарвовек) — генную терапию спинальной мышечной атрофии с биаллельной мутацией гена выживаемости моторных нейронов 1 (SMN1) у детей в возрасте младше двух лет. Препарат подходит как симптоматическим пациентам, так и предсимптоматическим, выявляемым генетическим тестированием. Регуляторное разрешение выдано «Авексис» (AveXis), которую « Новартис » (Novartis) купила за 8,7 млрд долларов. Онасемноген абепарвовек (onasemnogene abeparvovec, AVXS-101) представляет собой генотерапевтическое лечение на основе вектора AAV9, после единственной дозы обеспечивающее замену отсутствующего или дефектного гена SMN1 на его функциональную копию без изменения ДНК ребёнка . Итогом становится нормальная выработка белка SMN — и соответствующее прекращение прогрессирования спинальной мышечной атрофии. Во всяком случае однократное введение препарата Золгенсма демонстрирует , что пациенты начинают не только избавляться от зависимости в ИВЛ , но и демонстрировать существенное улучшение моторных навыков (способность сидеть, вставать, ходить), в ряде случаев полностью отвечающих нормальному возрастному развитию . Препарат считается наиболее дорогим лекарством со стоимостью курса (1 укол) более $2 млн . По некоторым оценкам, возможно порядка тысячи применений препарата до 2025 или 2027 года .
«Рош» (Roche) продолжает разработку рисдиплама , который, работая по аналогии с нусинерсеном, характеризуется существенным преимуществом, поскольку сделан в пероральной рецептуре .
Возможна только пассивная профилактика — консультирование родителей с риском СМА о возможных последствиях и пренатальная ДНК-диагностика во время беременности через биопсию ворсин хориона для принятия решения о рождении или прерывании беременности. Профилактика рождения больных детей возможна с помощью преконцепционного генетического тестирования с целью выявления носителей генетической мутации и преимплантационного генетического тестирования в цикле ЭКО для отбора эмбриона без мутаций, связанных со СМА.
Государственный регистр пациентов с диагнозом СМА не ведется. С 2016 года пациентов подсчитывает фонд «Семьи СМА». За это время удалось собрать информацию о 989 пациентах и 250—300 погибших. Теоретически (исходя из общемировой статистики заболеваемости) больных может быть около 2-3 тыс., считают в фонде .
По подсчетам фонда «Семьи СМА», в 2020 году лечением в России обеспечены лишь 129 больных.
В 2020 году единственным зарегистрированным в стране препаратом, показанным для терапии СМА является «Спинраза». Препарат «Спинраза» (« Нусинерсен ») в России был зарегистрирован в августе 2019 года. Его закупку должны оплачивать региональные власти, однако из-за дороговизны препарата лечение получают не все пациенты. Большинство пациентов, получающих лечение, добились его через суд. В регионах пациенты часто получают отказ в предоставлении лечения. Официально региональные власти объясняют это тем, что заболевание отсутствует в программе ВЗН (высокозатратных нозологий), в перечне жизнеугрожающих орфанных заболеваний или других целевых программах.
В августе 2020 года Миндзрав РФ принял решение о включении препарата в государственный перечень жизненно необходимых и важнейших лекарственных препаратов. Это означает фиксацию предельной отпускной цены на лекарство. Эта мера сделает лечение более доступным .
18 марта 2020 года швейцарская компания Roche («Рош») подала заявку на регистрацию в России препарата рисдиплам (торговое наименование «Эврисди»). В январе компания объявила о запуске глобальной программы дорегистрационного доступа к препарату рисдиплам для пациентов со СМА первого типа. Россия стала первой страной, в которой компания «Рош» открыла возможность участия в программе дорегистрационного доступа к препарату больных со СМА второго типа .
В середине июля 2020 года компания « Новартис » подала досье на регистрацию препарата «Золгенсма» в Министерство здравоохранения РФ. Пока «Золгенсма» не зарегистрирован для клинического применения в России, его ввоз на территорию страны может осуществляться только по жизненным показаниям для конкретных пациентов в соответствии с требованиями, определёнными федеральным законом № 61-Ф3 (об обращении лекарственных средств). Для этого необходимо решение врачебных комиссий медучреждений, а также одобрение на ввоз со стороны Министерства здравоохранения РФ.
Осуществлять применение этого препарата в России могут на сегодняшний день два федеральных центра, которые получили полномочия после прохождения необходимого обучения, организованного производителем «Золгенсма» — компанией АveXis (группа компаний «Новартис»). Оба учреждения являются ведущими российскими медицинскими центрами мирового уровня и имеют богатый опыт работы в педиатрии, включая лечение редких наследственных заболеваний .
На 23 июля 2020 года укол «Золгенсма» получили 16 российских пациентов . До 2021 года терапия «Золгенсмой» государством не финансировалась, все пациенты, прошедшие лечение, получали его за счет средств благотворительных фондов и отдельных меценатов. Известно, что на лечение детей с диагнозом СМА жертвовали средства бизнесмены Владимир Лисин , Владимир Гуриев , Алишер Усманов .
23 июня 2020 года президент России Владимир Путин выступил с инициативой повышения ставки НДФЛ с 13 % до 15 % для граждан, получающих более 5 млн рублей в год. По предварительным оценкам, эта мера даст бюджету порядка 60 млрд рублей, которые направят на лечение детей с орфанными заболеваниями . Соответствующий законопроект был одобрен Госдумой в первом чтении 21 октября 2020 года. Новый механизм помощи детям с тяжелыми редкими заболеваниями заработал с 1 января 2021 года , когда президентом был учрежден благотворительный фонд помощи больным детям « Круг добра ». К сентябрю 2021 года фонд одобрил финансирование лечения 9 пациентов со СМА «Золгенсмой» из федерального бюджета .
24 ноября 2020 года Правительство России включило препарат «Спинраза» в перечень жизненно необходимых и важнейших лекарственных препаратов. Распоряжение вступило в силу 1 января 2021 года .
Летом 2022 года Центр развития перспективных технологий (оператор системы маркировки « Честный знак ») сообщил, что «Золгенсма» официально введена в оборот лекарственных средств в России .
Кроме спинальных амиотрофий, обусловленных мутацией в генах SMN1 или SMN2 на длинном плече 5-й хромосомы (5q13.2), вызывающих поражение проксимальных мышц, существует множество схожих заболеваний, большинство из которых — с преимущественным поражением дистальных (то есть ближе к свободному концу конечности) мышц.
| Наименование и синонимы | OMIM | Ген | Локус | Тип наследования | Описание |
| X-сцепленная Спинальная амиотрофия 1-го типа (SMAX1), Spinal and bulbar muscular atrophy (SBMA), Kennedy’s disease (KD) | NR3C4 | Xq12 | Х-сцепленный, рецессивный | Позднее начало (в 40-60 лет), медленное прогрессирование, участие в процессе бульбарной группы черепных нервов, нисходящее распространение параличей | |
| X-сцепленная Спинальная амиотрофия 2-го типа (SMAX2), Arthrogryposis multiplex congenita — X-linked type 1 (AMCX1) | UBA1 | Xp11.23 | Х-сцепленный, рецессивный | Врождённая гипотония и арефлексия вследствие дегенерации и потери двигательных нейронов передних рогов спинного мозга и ствола головного мозга. Часто сочетается с врождёнными контрактурами и/или переломами. Интеллектуальное развитие нормальное. Заболевание быстро прогрессирует, приводя к смерти пациентов до 3-месячного возраста. | |
| X-сцепленная Спинальная амиотрофия 3-го типа (SMAX3), Distal spinal muscular atrophy — X-linked (DSMAX) | ATP7A | Xq21.1 | Х-сцепленный, рецессивный | Поражены дистальные мышцы всех конечностей, почти всегда у мальчиков, медленно прогрессирующее. | |
| Дистальная Спинальная амиотрофия (DSMA1), Spinal muscular atrophy with respiratory distress type 1 (SMARD1), Distal hereditary motor neuropathy type 6 (HMN6) | IGHMBP2 | 11q13.3 | Аутосомно-рецессивный | Признаки проявляются с самого рождения, реже во внутриутробном периоде. Заболевание характеризуется преимущественным поражением мышц верхних конечностей и развитием тяжёлых респираторных осложнений из-за прогрессирующей дегенерации мотонейронов передних рогов спинного мозга. | |
| Дистальная Спинальная амиотрофия 2-го типа (DSMA2), Distal hereditary motor neuropathy — Jerash type (HMN-J) | ? | 9p21.1-p12 | Аутосомно-рецессивный | Медленно прогрессирующее, описано только в одной семье | |
| Дистальная Спинальная амиотрофия 3-го типа (DSMA3), Distal hereditary motor neuropathy types 3 & 4 (HMN3, HMN4) | ? | 11q13.3 | Аутосомно-рецессивный | Медленно прогрессирующее | |
| Дистальная Спинальная амиотрофия 4-го типа (DSMA4) | PLEKHG5 | 1p36.31 | Аутосомно-рецессивный | Медленно прогрессирующее, описано только в одной семье | |
| Дистальная Спинальная амиотрофия 5-го типа (DSMA5) | DNAJB2 | 2q35 | Аутосомно-рецессивный | Начинается в молодом взрослом возрасте, медленно прогрессирующее. | |
| Дистальная Спинальная амиотрофия VA-типа (DSMAVA), Distal hereditary motor neuropathy type 5A (HMN5A) | GARS | 7p14.3 | Аутосомно-доминантный | Преобладает поражение верхних конечностей. | |
| Дистальная Спинальная амиотрофия VB-типа (DSMAVB), Distal hereditary motor neuropathy type 5B (HMN5B) | REEP1 | 2p11 | Аутосомно-доминантный | Преобладает поражение верхних конечностей. | |
| Дистальная Спинальная амиотрофия с преимущественным поражением голеней, Distal hereditary motor neuropathy type 2D (HMN2D) | FBXO38 | 5q32 | Аутосомно-доминантный | Проявляется в юношестве или у взрослых, медленно прогрессирует, поражает проксимальные и дистальные мышцы, сначала проявляется слабость в голенях, которая распространяется и на руки. | |
| Дистальная спинальная амиотрофия с преимущественным поражением голосовых связок, Distal hereditary motor neuropathy type 7A (HMN7A), Harper-Young myopathy. | SLC5A7 | 2q12.3 | Аутосомно-доминантный | Проявляется у взрослых параличом голосовых связок, очень редкое заболевание. | |
| Аутосомно-доминантная Спинальная амиотрофия, Distal hereditary motor neuropathy type 2A (HMN2A) | HSPB8 | 12q24.23 | Аутосомно-доминантный | Проявляется у взрослых. Аллельный вариант болезни Шарко-Мари-Тутса (CMT2L) | |
| Аутосомно-доминантная ювенильная Спинальная амиотрофия, Distal hereditary motor neuropathy type 1 (HMN1) | ? | 7q34-q36 | Аутосомно-доминантный | Проявляется в юном возрасте | |
| Врождённая дистальная Спинальная амиотрофия | TRPV4 | 12q24.11 | Аутосомно-доминантный | Поражение двигательных нейронов спинного мозга, иннервирующих нижнюю часть тела. Проявляется непрогрессирующей мышечной атрофией, атрофией мышц бёдер, мышц-разгибателей стопы, слабостью в коленях. Формируются контрактуры в коленных суставах и деформируются стопы. У некоторых пациентов может наблюдаться паралич голосовых связок. | |
| Лопаточно-малоберцовая Спинальная амиотрофия (SPSMA), Scapuloperoneal neurogenic amyotrophy | TRPV4 | 12q24.11 | Аутосомно-доминантный или | Поражает мышцы нижних конечностей. Очень редкое заболевание. Аллельный вариант врождённой дистальной спинальной амиотрофии. | |
| Ювенильная сегментальная Спинальная амиотрофия (JSSMA) | ? | 18q21.3 | ? | Начинается в юности, прогрессирует 2-4 года, после чего стабилизируется, влияет в первую очередь на руки, очень редкое. | |
| Спинальная амиотрофия Финкеля, Finkel-type proximal spinal muscular atrophy (SMA-FK) | VAPB | 20q13.32 | Аутосомно-доминантный | Средний возраст манифестации заболевания 37 лет (известны случаи в возрасте до 12 лет). Симметричная мышечная слабость и истощение мышц. Медленная потеря мышечной силы и прогрессирующая проксимальная атрофия, которая начинается в ногах и со временем распространяется на руки. Также у больных наблюдаются генерализованные фасцикуляции, гипоактивность или отсутствие глубоких сухожильных рефлексов. | |
| Спинальная амиотрофия Джокела, Jokela-type spinal muscular atrophy (SMA-J) | ? | 22q11.2-q13.2 | Аутосомно-доминантный | Позднее начало, медленно прогрессирование, поражает проксимальные и дистальные мышцы у взрослых. | |
| Спинальная амиотрофия с преимущественным поражением нижних конечностей 1, Spinal muscular atrophy with lower extremity predominance 1 (SMALED1) | DYNC1H1 | 14q32 | Аутосомно-доминантный | Поражает проксимальные мышцы у младенцев. | |
| Спинальная амиотрофия с преимущественным поражением нижних конечностей 2, Spinal muscular atrophy with lower extremity predominance 2 (SMALED2) | BICD2 | 9q22.31 | Аутосомно-доминантный | Врождённое или с ранним началом, поражающее преимущественно нижние конечности, не прогрессирует, очень редкое. | |
| Спинальная амиотрофия с прогрессирующей миоклонической эпилепсией, Spinal muscular atrophy with progressive myoclonic epilepsy (SMA-PME), Jankovic-Rivera syndrome. | ASAH1 | 8p22 | Аутосомно-рецессивный | Медленно прогрессирует, преимущественно поражает дистальные мышцы, сочетается с денервацией и миоклоническими приступами. | |
| Спинальная амиотрофия с атрофия с врожденными переломами костей, Spinal muscular atrophy with congenital bone fractures (SMA-CBF) | ? | ? | Аутосомно-рецессивный ? | Тяжёлое истощение мышц (как при болезни Верднига-Гоффмана), сопровождается врожденными переломами костей. | |
| Спинальная амиотрофия с понтоцеребеллярной гипоплазией, Spinal muscular atrophy with pontocerebellar hypoplasia (SMA-PCH), Pontocerebellar hypoplasia type 1A (PCH1A) | VRK1 | 14q32 | Аутосомно-доминантный | Описано восемь типов понтоцеребеллярной гипоплазии. Частота заболеваний неизвестна. Все формы заболевания имеют общие признаки: аномальное развитие головного мозга, проблемы с двигательной активностью, задержку развития, умственную неполноценность, прогрессирующую микроцефалию и церебральные проявления различной степени. Заболевание проявляется с рождения, в ряде случаев первые признаки отмечаются уже внутриутробно. Пациенты, как правило, погибают в раннем детском возрасте. | |
| Ювенильная асимметричная сегментальная Спинальная амиотрофия, Juvenile asymmetric segmental spinal muscular atrophy (JASSMA), Monomelic amyotrophy; Hirayama disease; Sobue disease | ? | ? | ? | Болезнь мотонейронов, которая поражает молодых (15-25-летних) мужчин в Индии и Японии. Начинается с мышечной атрофии, которая стабилизируется в плато после 2-5 лет, симптоматика не меняется. Нет боли или потери чувствительности. В отличие от других более низких мотонейронных болезней, MMA, как полагают, не наследуется и редко проявляется фасцикуляциями. |