Альтернирующие синдромы
- 1 year ago
- 0
- 0

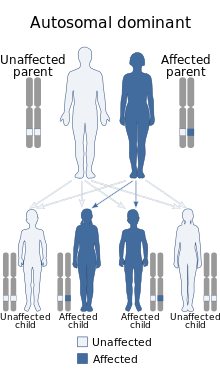
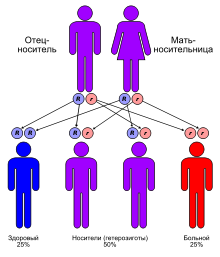
Синдром Макла — Уэльса (MWS) (иначе холодовой аутовоспалительный синдром ) — это мутация в гене CIAS1 с развитием холодового аутовоспалительного синдрома. Является редким аутовоспалительным заболеванием наследственного характера (к 1998 году было описано не более 100 случаев ). Преимущественно этническая распространённость — народы Северной Европы . Тип наследования — аутосомно-доминантный. Генетическая основа — мутация гена CIAS1, расположенного на длинном плече 1-й хромосомы (1q44) и кодирующего белка криопирина (пурин-подобный белок преимущественно экспрессирующийся в лейкоцитах периферической крови) . Данный белок является основной образуемого в клетке супрамолекулярного комплекса, называемого инфламмасомой , выполняющего функцию превращения pro-IL-1β (одного из основных провоспалительных цитокинов ) в активную форму, а также принимающего участие в выполнении программы апоптоза . Холодовой аутовоспалительный синдром тесно связан с двумя другими синдромами: семейной холодовой крапивницы и мультисистемным воспалительным заболеванием неонатального возраста — фактически, все связаны с мутациями гена. В целом Синдром Макла — Уэльса относится к группе (англ.) ((CAPS) .
Мутация в гене CIAS1 приводит к увеличению активности белка криопирин . Этот белок отвечает за реакцию организма на повреждение или инфекцию . В это время химическое вещество интерлейкин 1β вырабатывают иммунные клетки макрофаги. Это химическое вещество взаимодействует с рецептором на поверхности других клеток иммунной системы, они дают симптомы воспаления, такие как лихорадка, артрит и недомогание. Во время болезни, повышение активности криопирина приводит к увеличению интерлейкина 1β. Это приводит к воспалению во всём теле с соответствующими симптомами .
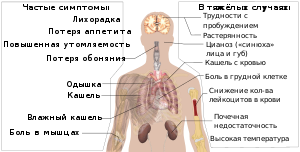
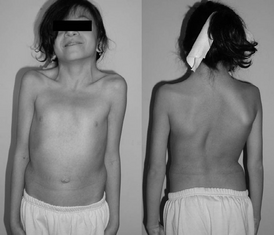
Первые симптомы появляются после переохлаждения и переутомления через 1,5 — 2 часа: сыпь, жар (лихорадка). В основном симптомы идут в такой последовательности:
Обычно смерть наступает из-за амилоидоза почек или остановки сердца.
Впервые болезнь была описана в 1962 году британскими исследователями ( англ. Thomas James Muckle) и ( англ. Michael Vernon Wells) .
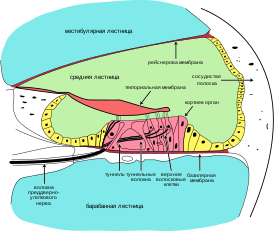
Макл и Уэльс описали семью из Дербишира , в которой крапивница, прогрессирующая перцептивная глухота и амилоидоз почек были объединены в преимущественно наследственный синдром. Новооткрытым заболеванием были затронуты пять поколений семьи. Вскрытие двух пациентов показало отсутствие Кортиева органа , атрофию улиткового нерва и амилоидную инфильтрацию почек . В 1969 году американский невролог ( англ. Joseph T. Black) описал пораженных болезнью лиц в трех поколениях одной семьи и отметил боли в конечностях ( артралгия ), как один из симптомов .
Ревматолог Бертело и соавторы в 1994 году исследовали четыре поколения семьи, в которой у семи членов проявились различные признаки синдрома, связанные с (щечной и генитальной молочницей) в пяти случаях и цистинурию в одном, двое других родственников страдали от ихтиоза . Никаких признаков амилоидоза обнаружено не было .
В 1996 году исследователи сообщили о трех поколениях семьи, в которой три сестры страдали интермиттирующей (эпизодической) крапивницей, полиартралгией и периодическими болями в животе. У все троих наблюдавшихся диагностирована микрогематурия, но никаких доказательств амилоидоза почек. Микроскопическая гематурия была установлена также у их матери и дочери одной из них. Ни один из пациентов не имел признаков глухоты .
Гербиг с соавторами в 1998 году описали 21-летняя женщину и её отца, страдавших от Синдрома Макла — Уэльса, у наблюдаемых задокументированы повышенные сывороточные уровни ИЛ-6 во время вспышки крапивницы. Симптомы и высокий уровень ИЛ-6 показали у дочери. Её крапивница преимущественно локализовалась на туловище и конечностях, была незудящая, и, казалось, следовала « внутренним часам » начинаясь после полудня, достигая кульминации к позднему вечеру, и затем исчезая в течение ночи. Все это часто сопровождалось лихорадкой, ознобами , спазмом кожных мышц, общим недомоганием и болями в конечностях, симптомы, как правило, чаще всего фиксировались по понедельникам. Утром не было абсолютно никаких симптомов. Пациент не может определенно выделить факторы приводящие к этому за исключением усталости и перегрева вызванного солнечной погодой. В возрасте 7 лет аудиограмма показала умеренную нейросенсорную тугоухость, но родители отказались от дальнейших исследований, поскольку отец, страдавший от аналогичных симптомов и показывавший постоянно повышенную скорость оседания эритроцитов , напротив чувствовал себя совершенно здоровым.
Исследовательская группа Гербига также заявила, что только около 100 случаев синдрома совмещающего симптомы крапивницы глухоты и амилоидоза были зарегистрированы после описания синдрома Маклом и Уэльсом в 1962 году у 9 членов семьи из Дербишира. Они предположили, что некоторые из спорадических случаев, в частности вероятно, были смешаны с другими расстройствами, к примеру, (хронический детский кожно-артикулярно-неврологический синдром) . Они также указали на спорадический случай, описанный Линке и соавторами в 1983 году в качестве вероятного примера к выдвинутой Маклом и Уэльсом конечной симптоматике синдрома .
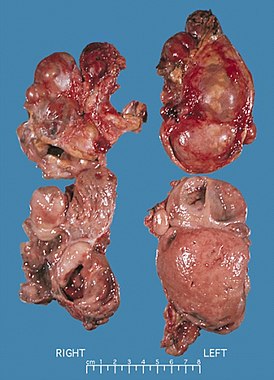
Либерман и соавторы описали гистологические особенности поражения кожи в спорадическом случае синдрома Макла — Уэльса. Так пациент, 54-летний мужчина, имел бессимптомные, затухающие очаги высыпаний начиная с возраста 2 месяцев, часто сопровождавшиеся ознобом, артралгией и на поздних стадиях — болями в нижних конечностях. Его детство было осложнено стойкими лимфаденопатией , гепатоспленомегалией , отеком диска зрительного нерва, анемией, «подагрическими» артритами и увеличением скорости оседания эритроцитов без видимой причины. В возрасте 19 лет перцептивная тугоухость привела к необходимости ношения двухсторонних слуховых аппаратов, кроме того был прописан курс антибиотиков против почечной инфекции. В период с 20 до 40 лет у него было несколько эпизодов культуро-отрицательного эозинофильного менингита с комой или судорогами. Установлено, что это была частичная реакция на кортикостероиды , и следовательно, исследователи предположили васкулит центральной нервной системы. показала хронический менингит с васкулярной астроглиальной реакцией без васкулита. В период с 30 до 50 лет он страдал рецидивными болями в животе, обильным поносом и нефротическим синдромом , требующим перитонеального диализа . Ректальная и почечная биопсии показали амилоидные отложения. В возрасте 50 лет ему была сделана двухсторонняя трансплантация роговицы по причине ленточной кератопатии (кальцификации роговицы). В возрасте 54 лет, несмотря на противовоспалительную терапию, в том числе преднизолоном , азатиоприном и циклоспорином , его состояние постепенно ухудшилось, и он умер во время гипотензивного эпизода. Семейный анамнез был отрицательным для того что бы сделать вывод о соответствии симптомам синдрома Макла — Уэльса. Диагноз синдром Макла — Уэльса был поставлен лишь незадолго до смерти пациента .
В клинической практике отмечаются приступы лихорадки, сопровождающиеся артралгией, уртикарной сыпью (без зуда), явлениями конъюнктивита, реже — микрогематурия, лимфаденопатия, гепатоспленомегалия, боли в животе. Иногда встречаются другие симптомы, характерные для родственных синдромов CAPS: реакция на холод, неврологические проявления (галлюцинации). У больных выявляют положительный тест ревматических розеток, увеличение содержания IgG и IgA , повышение СОЭ , лейкоцитоз. При лабораторном исследовании во время приступа определяется повышение С-реактивного белка , лейкоцитоз. Прогноз определяется глухотой, развивающейся вследствие атрофии слухового нерва, симптомами прогрессирующего мультиорганного амилоидоза АА типа.
В 14 серии 7 сезона телесериала « Доктор Хаус » с этим синдромом обратился пациент.