Interested Article - Реакция Дильса — Альдера

- 2020-01-07
- 1

Реакция Ди́льса — А́льдера (диеновый синтез) — реакция [4+2]-циклоприсоединения и сопряжённых диенов с образованием шестичленного цикла.
В реакцию вступают циклические и ациклические сопряжённые диены , енины −C=C−C≡C− или их гетероаналоги — соединения с фрагментами −С=С−С=О, −С=С−С≡N. Диенофилами обычно являются алкены и алкины с кратной связью, активированной электроноакцепторными заместителями. В роли диенофилов также могут выступать соединения, содержащие двойные связи с гетероатомом , например >С=О, >С=N−, −СN, −N=О, −S=O, −N=N− .
История
В 1902 году в лаборатории И. Тиле аспирантом В. Альбрехтом была проведена реакция между циклопентадиеном и бензохиноном с целью получения сопряжённого полиена, однако вместо ожидаемого продукта конденсации было получено два дикетона — продукты присоединения одной или двух молекул циклопентадиена по двойным связям хинона. Строение полученных веществ не было установлено, а реакция не получила дальнейшего развития в этой лаборатории .
Также протекание реакции Дильса — Альдера наблюдали в 1910 году С. В. Лебедев (при димеризации изопрена ), а в 1920 году — Г. фон Эйлер и К. Джозефсон (при взаимодействии изопрена с бензохиноном) .
Систематическое изучение реакции между 1,3- диенами и сопряжёнными кетонами было представлено Отто Дильсом и Куртом Альдером в 1928 году в журнале Justus Liebigs Annalen der Chemie . Авторы статьи привели свидетельства общего характера реакции, а также с уверенностью указали на возможность её использования в синтезе природных соединений, при этом заявив :
Мы со всей определённостью оставляем за собой права на использование открытой нами реакции для решения этих синтетических проблем.
В следующие за открытием годы были изучены основные закономерности протекания реакции и показана широкая область её применимости, а авторы открытия в 1950 году были удостоены Нобелевской премии по химии .
Механизм
Реакция Дильса — Альдера представляет собой согласованное [4+2]- циклоприсоединение , протекающее между 1,3- диеном и ненасыщенным соединением — диенофилом. Обычно диен содержит электронодонорный заместитель, а диенофил — электроноакцепторную группу. Менее распространён обращённый вариант, когда электронообогащённым соединением является диенофил .
С точки зрения теории граничных орбиталей , реакцию можно представить как взаимодействие высшей занятой молекулярной орбитали (ВЗМО) электронодонорного диена и низшей свободной молекулярной орбитали (НСМО) диенофила. В случае обращённого варианта взаимодействуют НСМО диена и ВЗМО диенофила. По этой причине изменение заместителей в реагентах противоположным образом влияет на протекание классического и обращённого вариантов реакции. Например, классическая реакция Дильса — Альдера ускоряется при увеличении донорной способности диена, а обращённая, напротив, замедляется .
Активность реагентов
Диены

Для участия в реакции [4+2]-циклоприсоединения диен принимает плоскую s-цис - конформацию , в которой обе двойные связи находятся по одну сторону от одинарной C–C-связи .
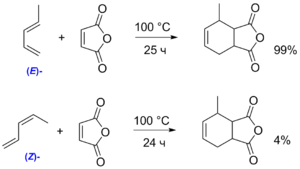
Одним из наиболее активных диенов является циклопентадиен-1,3 , в котором s-цис конформация закреплена. В случае бутадиена-1,3 скорость реакции заметно ниже, поскольку он существует в виде двух ротамеров ( s-цис и s-транс ). Однако энергия перехода между этими конформациями невелика, поэтому бутадиен может быть использован как диенофил в реакции Дильса — Альдера. 1-Алкилзамещённые диены вступают в реакцию с различной скоростью в зависимости от конфигурации двойной связи. Так, E - пиперилен достаточно активно реагирует с малеиновым ангидридом , тогда как для Z -изомера выход составляет лишь 4 % (через 24 часа при 100 °C). Подобное поведение объясняется невыгодностью s-цис -конформации для Z -пиперилена. В случае диенов, которые существуют в закреплённой s-транс -конформации, реакция Дильса — Альдера становится невозможной. Например, в отличие от активного α- фелландрена , β-фелландрен вовсе не вступает в циклоприсоединение с малеиновым ангидридом .
Диенофилы
Наиболее активными диенофилами являются алкены и алкины с электроноакцепторными заместителями (α,β-непредельные альдегиды , кетоны , карбоновые кислоты и их производные, винилсульфоны, нитроалкены). С увеличением числа акцепторных групп происходит увеличение активности диенофила. Так, в реакции с циклопентадиеном 1,1-дицианэтилен в 4,5·10 4 раз более активен, чем акрилонитрил (моноцианэтилен). Тетрацианэтилен ещё более активен и поэтому часто используется на практике в качестве «ловушки» генерируемых in situ молекул с 1,3-диеновым фрагментом .
Часто в качестве диенофилов используются дизамещённые алкены и алкины, например, малеиновая кислота , её эфиры, ацетилендикарбоновая кислота , п -бензохинон и другие соединения. В реакцию Дильса — Альдера вводят также непредельные углеводороды, хоть они и гораздо менее активны и реагируют только при нагревании. В частности, реакции этилена и ацетилена с циклопентадиеном могут применяться для синтеза и норборнадиена .
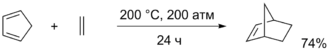
Для синтеза бензаннелированных соединений в качестве диенофила применяется , получаемый in situ из о -бромфторбензола под действием магния или диазотированием антраниловой кислоты .
Региоселективность реакции
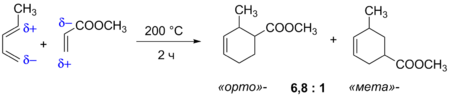
При введении в реакцию Дильса — Альдера несимметричных реагентов наблюдается региоселективное образование продуктов циклоприсоединения. Так, при реакции 1-замещённых диенов с несимметричными алкенами преимущественно образуются продукты, в которых заместители расположены у соседних атомов углерода (так называемые « орто »-продукты), а продукты с 1,3-замещением (« мета »-продукты) получаются в меньшем количестве. Эта закономерность наблюдается для ряда заместителей в молекуле диена, а также различных диенофилов. Для её объяснения можно рассматривать возникающее под действием заместителей распределение зарядов в молекулах реагентов. Согласно такой модели, избирательность реакции должна возрастать с увеличением донорной способности заместителя в диене и акцепторной способности заместителя в диенофиле .
В реакциях 2-замещённых диенов общей закономерностью является образование « пара »-продуктов, которое также может быть объяснено на основании поляризации реагентов .
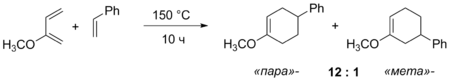
В случае дизамещённых и полизамещённых диенов ситуация усложняется и реакции протекают менее селективно и предсказуемо .
Стереохимия реакции
Поскольку реакция Дильса — Альдера протекает как согласованный процесс через циклическое переходное состояние , конфигурация продукта реакции определяется конфигурацией исходных реагентов. Так, из ( Z )-алкенов образуются цис -продукты, а из ( E )-алкенов — транс -продукты . Подобный принцип применим и для заместителей в 1 и 4 положениях диена: если конфигурации двух двойных связей диена совпадают, то в продукте эти заместители находятся в цис -расположении .
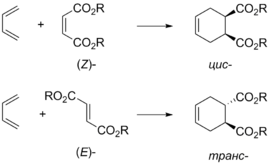
|
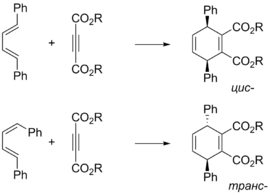
|
Было замечено, что при реакции циклических диенов с различными диенофилами образуется, в основном, один из двух возможных диастереомеров с эндо -расположением заместителей диенофила. Соотношение эндо - и экзо -изомеров может принимать различные значения (от 3:2 до 20:1). Данная закономерность называется эндо -правилом Альдера. Эндо -правило выполняется и для реакций с участием ациклических 1,4-дизамещённых диенов. Наблюдаемые результаты объясняются вторичными орбитальными взаимодействиями, которые возникают при сближении акцепторной группы диенофила с С2- и С3-атомами диена. Данные взаимодействия стабилизируют переходное состояние и способствуют образованию эндо -продукта .
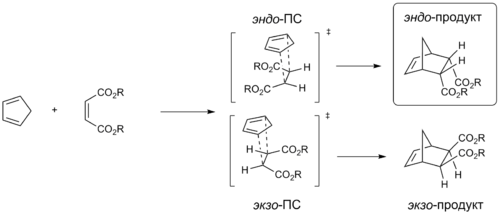
Катализ кислотами Льюиса
Долгое время считалось, что на реакцию не действуют какие-либо катализаторы , однако в 1960 году было показано, что реакция между антраценом и малеиновым ангидридом значительно ускоряется в присутствии хлорида алюминия AlCl 3 . В присутствии катализатора она протекает мгновенно при комнатной температуре, тогда как в некаталитическом варианте требуется кипячение в ксилоле (140 °C) в течение 72 часов. Кроме того, применение катализатора повышает региоселективность реакции и соотношение эндо - и экзо -изомеров .
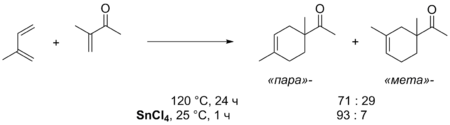
Действие кислот Льюиса как катализаторов объясняется их способностью образовывать комплексы с диенофилами, за счёт чего происходит понижение энергии НСМО диенофила и уменьшение разницы энергий взаимодействующих НСМО диенофила и ВЗМО диена. Данный факт приводит к резкому увеличению скорости реакции . Влияние катализатора на селективность реакции наблюдается из-за изменения величин орбитальных коэффициентов в диенофиле и возрастающего различия в силе орбитальных взаимодействий при реализации альтернативных переходных состояний, приводящих к изомерным продуктам .
Благодаря своим преимуществам, каталитическая реакция Дильса — Альдера широко используется в лабораторном синтезе различных органических соединений .
Стереоселективная реакция Дильса — Альдера
Первые исследования стереоселективных вариантов реакции Дильса — Альдера были основаны на использовании хиральных диенофилов, в частности, сложных эфиров непредельных карбоновых кислот с хиральными спиртами . При этом было обнаружено, что на стереоселективность реакции сильно влияют условия её проведения. Например, реакция между бутадиеном и (–)-диметилфумаратом при нагревании протекала с низкой стереоселективностью, а использование катализатора AlCl 3 приводило к получению продукта с 72—76 % .
Продуктивным подходом в данной области оказалось использование оксазолидиновой методологии Эванса, в рамках которой в качестве диенофилов выступали α,β-ненасыщенные N -ацилоксазолидиноны. Согласно предложенной модели, данные реагенты образуют хелаты с кислотой Льюиса ((C 2 H 5 ) 2 AlCl), в которых одна из сторон пространственно блокируется заместителем в оксазолидиноне, что и определяет стереоселективность реакции. Кроме того, получаемые продукты можно разлагать с регенерацией оксазолидиноновых фрагментов. Этот и подобные подходы, в которых диены и диенофилы содержали удаляемые вспомогательные хиральные группы, были использованы в синтезе многих природных соединений .
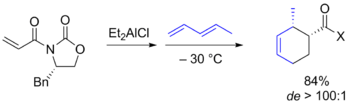
Позже была показана эффективность использования хиральных кислот Льюиса в качестве асимметрических индукторов. Одним из эффективных катализаторов оказался ( R , R )-дихлор-2-нафтилциклогексилборан. Его применение в реакции циклопентадиена и метиловых эфиров акриловой , кротоновой или фумаровой кислот приводит к продукту с энантиомерным избытком 86—97 %. Широкое исследование было посвящено использованию хиральных , синтезируемых из природных аминокислот .
В биохимии
Реакции [4+2]-циклоприсоединения встречаются во вторичном метаболизме разнообразных организмов. Протекающие в живой природе реакции Дильса — Альдера относятся к процессам биосинтеза поликетидов , изопреноидов , фенилпропаноидов , а также алкалоидов и других продуктов смешанного биосинтеза. Катализирующие эти реакции ферменты — дильсальдеразы — представляют собой белки или РНК . Возможность ферментативного катализа для реакций этого типа представляется весьма интригующим аспектом, так как высокое сходство структуры переходного состояния и продукта реакции ( аддукта ) предполагает конкурентное ингибирование последним специфического фермента, катализирующего реакцию. И действительно, полученные моноклональные антитела , проявляющие высокоспецифичную дильсальдеразную активность, как и ожидалось, ингибируются продуктом реакции. Катализ же рибозимами низкоспецифичен, необходимой частью активного рибозима является координированный ион переходного металла , поэтому считается, что катализ рибонуклеиновой кислотой подобен катализу кислотой Льюиса. Предполагается, что изучение биосинтетических реакций Дильса — Альдера может привести к открытию новых механизмов ферментативного катализа. Возможно, вступающие в реакцию диеновый компонент и диенофил дестабилизируются , что способствует понижению энергии активации и эффективному протеканию реакции в физиологических условиях .
В настоящее время дильсальдеразы представляют большой интерес и активно изучаются.
Применение
Реакция используется для получения полициклических соединений, в том числе стероидов . С помощью реакции, также возможно получение многих хлорорганических пестицидов циклодиенового ряда: альдрина и его производных, гептахлора , , хлордана , мирекса и т.д. .
См.также
Примечания
- Химическая энциклопедия. В пяти томах / Гл. ред. И. Л. Кнунянц. — Советская энциклопедия, 1990. — Т. 2. — С. 54—55. — ISBN 5-85270-035-5 .
- ↑ , с. 605—607.
- ↑ , с. 607—609.
- ↑ , с. 609—612.
- ↑ , с. 612—618.
- Зауэр Е. // Успехи химии. — 1969. — Т. 38 , № 4 . — С. 624—661 .
- ↑ , с. 618—621.
- ↑ , с. 621—631.
- Emily M. Stocking and Robert M. Williams. Chemistry and biology of biosynthetic Diels–Alder reactions. (Review) (англ.) // Angewandte Chemie International Edition : Научный журнал. — 2003. — Vol. 42 , no. 27 . — P. 3078—3115 . — doi : . — .
- Вредные вещества в промышленности. Справочник для химиков, инженеров и врачей. Изд. 7-е, пер. и доп. В трех томах. Том I. Органические вещества. Под ред. засл. деят. науки проф. Н. В. Лазарева и докт. мед. наук Э. Н. Левиной. Л., «Химия», 1976. 592 стр., 27 табл., библиография —1850 названий.
Литература
- Оригинальные работы
- Diels O, Alder K. Synthesen in der hydroaromatischen Reihe (нем.) // Justus Liebigs Annalen der Chemie. — 1928. — Bd. 460 , Nr. 1 . — S. 98—122 . — doi : .
- Русскоязычные источники
- Смит В. А., Дильман А. Д. Глава 22. Реакция Дильса — Альдера. Часть I // Основы современного органического синтеза. — М. : Бином. Лаборатория знаний, 2009. — С. 605—642. — ISBN 978-5-94774-941-0 .
- Смит В. А., Дильман А. Д. Глава 23. Реакция Дильса — Альдера. Часть II // Основы современного органического синтеза. — М. : Бином. Лаборатория знаний, 2009. — С. 643—681. — ISBN 978-5-94774-941-0 .
- 2020-01-07
- 1