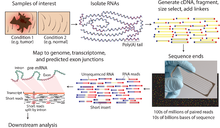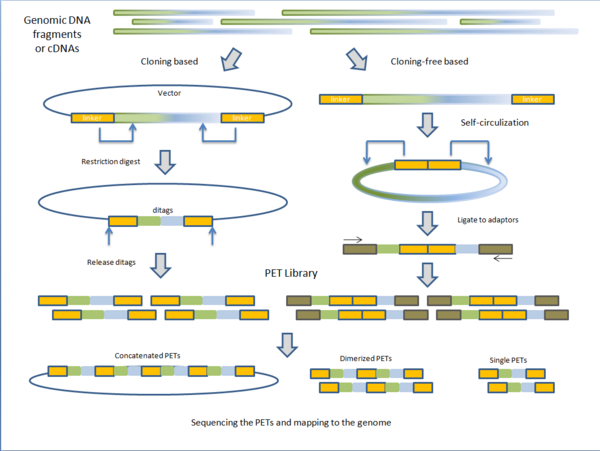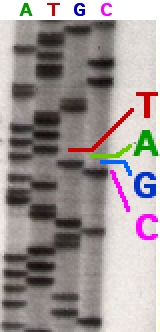
Секвенирование древней ДНК
- 1 year ago
- 0
- 0


Секвени́рование экзо́ма ( англ. Exome sequencing ) — секвенирование всех белок -кодирующих генов в геноме (то есть экзома ). Под секвенированием экзома подразумеваются две операции: во-первых, отбор экзонов . В зависимости от организма экзоны покрывают 1—2 % генома . У человека их насчитывается около 180 000, примерно 1 % от всего генома , или приблизительно 30 миллионов пар оснований (п. о.). Во-вторых, секвенирование экзонов с использованием любой платформы высокопроизводительного секвенирования ДНК и анализ полученных результатов .
Секвенирование экзома позволяет обнаружить генетические изменения, приводящие к изменению белковых последовательностей, которые могут в свою очередь приводить к возникновению заболеваний, таких как атеросклероз , болезнь Альцгеймера и других. Основное преимущество экзомного секвенирования заключается в возможности проводить массовый скрининг генов и обнаруживать мутации , ассоциированные с заболеваниями, при этом данная процедура оказывается проще и дешевле, чем .

Секвенирование экзома включает в себя четыре этапа: из предоставленного материала, отбор интересующей фракции ДНК (обогащение образцов), секвенирование отобранного материала и анализ полученных результатов .
Первый этап заключается в подготовке высококачественных препаратов геномной ДНК из предоставленных образцов путем отделения ДНК от белков , липидов и т.д. Стандартный метод — экстракция смесью фенол-хлороформ .
Стратегии обогащения образцов позволяют селективно отбирать нужные геномные участки, то есть экзоны, из образцов ДНК до этапа секвенирования. С момента описания первого оригинального метода в 2005 году разработано несколько стратегий обогащения образцов, подходящих для целей экзомного секвенирования . Выбор конкретного метода зависит от размеров интересующих участков, потребности в покрытии при секвенировании, имеющегося в наличии оборудования и других причин .
Полимеразная цепная реакция (ПЦР) широко применяется для амплификации требуемых фрагментов ДНК уже более 20 лет . Обычно в ПЦР используют только 2 праймера , однако разработаны методы , которая использует несколько праймеров и позволяет одновременно амплифицировать несколько ДНК-мишеней в ходе одного процесса. Подходы, использующие ПЦР, очень эффективны, но не позволяют работать с участками генома длиной в несколько миллионов п.о. из-за высокой цены и низкого качества получающихся образцов .

Метод — это техника, которая позволяет получить образцы ДНК, обогащенные амплифицированными инвертированными участками целевых последовательностей . Отбор нужных последовательностей происходит за счет замыкания интересующего участка в кольцо. Праймер здесь представляет собой одноцепочечный ДНК- олигонуклеотид , в центральной части которого содержится универсальная последовательность с сайтами рестрикции , а концы комплементарны двум участкам геномной ДНК, между которыми находится интересующая последовательность. Образцы, не вступившие в реакцию, остаются линейными и удаляются экзонуклеазами . Метод может быть полезен для работы с небольшим числом мишеней в большом количестве образцов. Главный недостаток — единообразие получаемых образцов, а также высокая цена при необходимости покрыть большой набор участков .
Для гибридизационного обогащения образцов экзомными участками создаются специальные микрочипы , содержащие закрепленные на подложке одноцепочечные олигонуклеотиды ( зонды ) с последовательностями из генома, способными покрыть интересующие участки. Геномная ДНК разрезается на фрагменты. Концы фрагментов делают с помощью рестриктаз , добавляют с универсальными праймерами. После гибридизации фрагментов с зондами на микрочипах негибридизованные фрагменты отмываются с подложки, а оставшиеся затем амплифицируются с помощью ПЦР . Ограничения метода связаны с дороговизной аппаратуры, количеством зондов, которые можно разместить на матрице, и необходимостью достаточно больших количеств ДНК для анализа .

В растворе синтезируется набор зондов, которые фиксируются на стрептавидиновых шариках. Шарики помещаются в раствор с фрагментированной геномной ДНК, где происходит селективная гибридизация зондов с нужными геномными участками, после чего шарики с интересующими фрагментами осаждают и отмывают. Затем оставшиеся участки секвенируют. Этот метод был разработан для усовершенствования метода гибридизационного обогащения: он позволяет создать избыток зондов к целевым участкам по сравнению с необходимым количеством образца. Оптимальный размер целевого участка ДНК — около 3,5 миллиона п.о., так при последующем секвенировании получается хорошее .
Основными поставщиками платформ для обогащения экзома являются , Agilent и Illumina .
| NimbleGen’s SeqCap EZ Exome Library | Agilent’s Sure Select Human All Exon Kit | Illumina’s TruSeq Exome Enrichment Kit | Illumina’s Nextera Rapid Capture Exome Kit | |
|---|---|---|---|---|
| Длина зондов | 55 — 105 | 114 — 126 | 95 | 95 |
| Рекомендованное количество ДНК пробы | 3 мкг | 3 мкг | 500 нг | 50 нг |
| Тип нуклеиновой кислоты зонда | ДНК | РНК | ДНК | ДНК |
| Стратегия покрытия интересующего фрагмента зондами | Перекрывающиеся зонды | Чаще строго последовательные зонды, чем перекрывающиеся | Гэпы между последовательностями зондов (зонды находятся на некотором расстоянии друг от друга по последовательности фрагмента) | Гэпы между последовательностями зондов |
| Метод фрагментации | Ультразвук | Ультразвук | Ультразвук | Транспозаза |
| Размер целевого фрагмента (для человека) | 64 | 50 | 62 | 62 |
| , остающиеся после фильтрации | 66 % | 71,7 % | 54,8 % | 40,1% |
| Основные сильные стороны | Высокая чувствительность и специфичность. Наиболее равномерное покрытие в трудных регионах . | Хорошее покрытие . Высокая скорость выравнивания . Меньше повторных прочтений, чем у других платформ . | Хорошее покрытие нетранслируемых областей и микроРНК | Хорошее покрытие нетранслируемых областей и микроРНК |
| Основные слабые стороны | Больше повторных чтений, чем у Agilent. Меньшая скорость выравнивания. | Меньше качественных чтений, чем у NimbleGen | Высокий уровень нецелевого обогащения | Высокий уровень нецелевого обогащения. Смещение покрытия для областей с высоким GC-составом , снижающим однородность. |
| Использование не только для человеческих последовательностей | Да | Да | Нет | Нет |
В настоящее время, в дополнение к наборам, нацеленным лишь на человека, NimbleGen предлагает наборы для экзомов кукурузы , ячменя , пшеницы , сои , мыши и свиньи , а Agilent — для экзомов мыши, крупного рогатого скота и рыбок данио-рерио . Оба поставщика также предлагают возможность разрабатывать индивидуальные комплекты для других видов. Наборы для нечеловеческих видов используют протоколы и зонды, аналогичные человеческим наборам поставщиков. Оба производителя предлагают гибкий процесс проектирования, который позволяет вносить изменения для улучшения покрытия для конкретных регионов и целей .
Существует несколько технологий секвенирования, включая классический метод секвенирования по Сэнгеру . Методы секвенирования нового поколения используют платформы Illumina , SOLiD и Ion-Torrent . Все эти методы могут использоваться в том числе и для секвенирования экзома .
Первичные данные секвенирования представляют собой огромный набор небольших последовательностей (чтений), длина и качество которых зависят от технических характеристик секвенатора и способа приготовления образцов. Качество чтений можно контролировать, например, при помощи программного пакета FastQС . Полученные чтения фильтруются: отрезаются концевые участки, которые часто имеют большое число ошибок, удаляются адаптерные последовательности (например, с помощью Trimmomatic или sickle ); затем корректируются ошибки (например, с помощью программ Bloocoo и Lighter ). Отфильтрованные чтения картируются на геном, где собираются в последовательности, соответствующие экзонам. В настоящий момент существует множество программ, которые осуществляют каждый этап подготовки данных секвенирования и их анализа, большинство из них требует больших вычислительных мощностей , так как объём получаемых данных очень велик .
Используя экзомное секвенирование, в ходе исследований с фиксированными затратами мы можем секвенировать последовательности с существенно большей глубиной покрытия по сравнению с покрытием, получаемым методами полногеномного секвенирования. Благодаря этому экзомное секвенирование чаще используется при решении задач, требующих надежного определения однонуклеотидных полиморфизмов .
29 сентября 2011 года компания Ambry Genetics стала первой сертифицированной компанией, предлагающей секвенирование экзома и диагностику заболеваний на его основе . В компании утверждают, что результаты экзомного секвенирования позволят сотрудникам диагностировать заболевания, при которых традиционные диагностические подходы неприменимы .
Идентификация мутаций, вызывающих заболевания, может внести существенный вклад в диагностические и терапевтические подходы, поможет прогнозировать развитие заболевания и позволит тестировать родственников, находящихся в зоне риска . Есть несколько факторов, на основании которых экзомному секвенированию отдается предпочтение перед моногенным анализом: возможность идентифицировать мутации в генах, не подвергшихся тестированию ввиду нетипичного клинического проявления и идентификация клинических случаев, при которых мутации в разных генах вызывают различные проявления у одного и того же пациента . Кроме того, метод позволяет диагностировать заболевания на ранних этапах и у молодых пациентов до проявления всего спектра характерных симптомов; он также используется для пренатальной диагностики В некоторых случаях пренатальное секвенирование экзома позволяет выявить генетические заболевания , в то время как стандартные методы ( кариотипирование и использование микрочипов) оказываются неэффективны .
Авторы важнейшей рецензированной публикации об экзомном секвенировании подчеркивают полезность этого метода для клинической практики. Авторы, применившие экзомное секвенирование для идентификации мутации, вызывающей синдром Барттера и , заявляют: «Мы видим будущее, в котором подобная информация станет частью повседневной клинической оценки пациентов с подозрениями на генетические заболевания с неясным диагнозом… Мы предвидим, что полноэкзомное секвенирование внесет огромный вклад в понимание того, какие гены и какими путями участвуют в развитии редких и частых человеческих болезней, а также в клиническую практику» .
Текущие крупные международные исследования направлены на определение в геноме частых полиморфизмов, которые легче всего идентифицировать современными методами. Однако из-за отрицательного отбора полиморфизмы, вызывающие крайне тяжелые заболевания, в частности, менделевские болезни, встречаются с существенно меньшей аллельной частотой и могут остаться невыявленными в ходе поиска генов-кандидатов при использовании современных стандартных методов * , при этом чаще всего они расположены в границах экзома. Так как при комплексных расстройствах с риском заболевания связано большое количество генов, для обнаружения их необходимы исследования очень большой выборки, поэтому, с точки зрения издержек, полногеномное секвенирование не является оптимальным. К тому же, полиморфизмы в кодирующих областях изучаются очень подробно, и их функциональное значение проще определить Успешная модель идентификации менделевских генов включает определение возникающих полиморфизмов при секвенировании генов двух родителей и потомка .
Геномы растений могут быть чрезвычайно сложными, повторяющимися и часто полиплоидными ; в результате некоторые из наиболее экономически важных культур не удается исследовать с использованием полногеномного секвенирования. Разработан набор для обогащения экзома пшеницы на основе накопленных данных транскриптома , с использованием которого были проведены исследования нежелательной внутрикультурной экзома, влияющей на фенотип растения, в частности, скорость роста, способность жить в различных условиях и другие важные для селекции признаки. Подобные же наборы были использованы при исследовании риса Oryza sativa и сои Glycine max . Также можно идентифицировать генетические маркеры , отвечающие за особую устойчивость растительных культур к определенным патогенам .
В ряде случаев секвенирование экзома может быть использовано как альтернатива более дорогому секвенированию полного генома, например, при исследовании генетических вариаций внутри и между популяциями .
Методы микрочипирования требуют зондов для гибридизации с известной последовательностью, поэтому они ограничены требованиями к разработке зондов и не позволяют выявить некоторые генетические изменения. Технологии высокопроизводительного секвенирования, используемые для секвенирования экзома, позволяют узнать последовательности гораздо большего числа локусов одновременно и определить неизвестные до сих пор источники многих болезней , то есть позволяют обойти ограничения генотипирующих чипов и классического секвенирования .
Секвенирование экзома — процедура более дорогая, но по мере уменьшения финансовых затрат и увеличения производительности методов секвенирования этот метод все шире используется в практике для диагностики редких генетических заболеваний .
Некоторые болезни могут быть связаны с мутациями в некодирующих областях или структурными перестройками, которые секвенирование экзома не позволит выявить . Но из-за дороговизны полногеномного секвенирования на нынешнем этапе развития науки и технологий экзомное секвенирование представляется оптимальным методом для клинической диагностики редких наследственных заболеваний, не выявляемых микрочипами .
Статистический анализ больших объёмов данных в ходе секвенирования экзома — отдельная трудоемкая задача. Есть несколько подходов для улучшения качества экзомных данных :
Для некоторых биологических видов качество сборки генома и его значительно хуже, чем для человека (или секвенированного генома нет вовсе). Это существенно ограничивает применение секвенирования экзома к другим организмам, поскольку осложняет обогащение образцов ДНК и картирование результатов секвенирования на геном .