Синдром Экарди
- 1 year ago
- 0
- 0

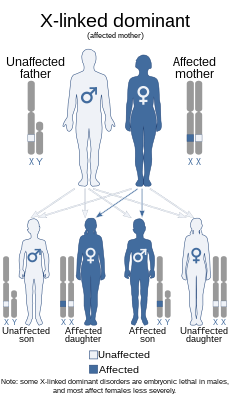
Синдро́м Экарди́ — редкий генетический порок развития , характеризующийся частичным или полным отсутствием одной из основных структур мозга — мозолистого тела , нарушением структуры сетчатки и инфантильными спазмами . Предположительно, синдром Экарди вызывается дефектом в Х-хромосоме , так как до сих пор наблюдался лишь у мальчиков с синдромом Клайнфельтера . Симптомы обычно возникают у детей в возрасте до 5 месяцев.
Это заболевание было впервые выделено в качестве отдельного синдрома в 1965 году Жаном Экарди , французским невропатологом .
Почти все описанные случаи синдрома Экарди были обнаружены у женщин. У нескольких мужчин с подтвержденным синдромом Экарди, была выявлена мутация XXY по 47-й хромосоме, называемая синдромом Клайнфельтера . Для мужчин, имеющих только одну X-хромосому, синдром Экарди летален.
У детей чаще всего выявляют синдром Экарди в возрасте до пяти месяцев. Значительное число девочек появляются в результате нормальных родов и, по-видимому, нормально развиваются примерно до трехмесячного возраста, когда у них начинаются детские спазмы. Начало инфантильных спазмов в этом возрасте связано с закрытием окончательных нейронных синапсов в головном мозге-стадии нормального развития мозга. Сообщалось о ряде опухолей, связанных с синдромом Экарди: папиллома сосудистого сплетения (наиболее распространенная), медуллобластома , гиперпластические полипы желудка, ректальные полипы, доброкачественная тератома мягкого неба, гепатобластома, эмбрионально-клеточный рак парафаринги, ангиосаркома конечностей и липома скальпа.
Синдром Экарди обычно характеризуется следующей триадой признаков — однако отсутствие одного из «классических» признаков не исключает диагноза синдрома Экарди, если присутствуют другие подтверждающие признаки.
| Пороки ЦНС | |
|---|---|
|
Синдромы,
с нарушением развития ЦНС |
|