Постоянная тонкой структуры
- 1 year ago
- 0
- 0

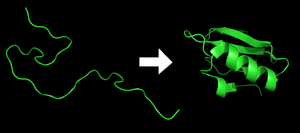
Предсказа́ние структу́ры белка́ ( англ. protein structure prediction ) — направление молекулярного моделирования , предсказание по аминокислотной последовательности трёхмерной структуры белка ( вторичной , третичной или четвертичной ). Данная задача является одной из самых важных целей биоинформатики и теоретической химии . Данные, полученные при помощи предсказания, применяются в медицине (например, в фармацевтике ) и биотехнологии при создании новых ферментов ).
Огромные объёмы данных о последовательности белков стали доступны в результате современных широкомасштабных работ по секвенированию ДНК , таких как проект «Геном человека» . Несмотря на усилия всего сообщества в области структурной геномики , количество экспериментально определённых белковых структур — обычно с помощью трудоёмкой и относительно дорогой рентгеновской кристаллографии или ЯМР-спектроскопии — значительно отстаёт от количества белковых последовательностей , что делает предсказание третичной структуры белка крайне востребованным .
Предсказание структуры белка остаётся чрезвычайно трудной и не до конца разрешённой задачей. Две основные проблемы — это расчёт свободной энергии и нахождение глобального минимума этой энергии . Метод предсказания структуры белка должен исследовать пространство всех возможных структур белка, которое является астрономически большим. Эти проблемы можно частично обойти с помощью и методах распознавания укладки (фолда) , в которых пространство поиска сокращается из-за предположения, что рассматриваемый белок принимает структуру, близкую к экспериментально определённой структуре другого гомологичного белка. С другой стороны, методы предсказания структуры белка ab initio должны явно разрешать эти проблемы, не опираясь на начальные предположения .
В декабре 2020 года команда DeepMind (исследовательского подразделения Google ) объявила о решении фундаментальной научной проблемы предсказания структуры белка. Программа, разработанная компанией и основанная на нейросетях, смогла предсказывать структуру белка с высокой точностью.

Альфа-спираль является наиболее распространённым типом вторичной структуры в белках. Альфа-спираль имеет 3,6 аминокислоты на поворот, а Н-связь образуется между каждым четвёртым остатком; средняя длина составляет 10 аминокислот (3 витка) или 10 Å , но варьируется от 5 до 40 (от 1,5 до 11 витков). Выравнивание Н-связей создаёт дипольный момент для спирали с результирующим частичным положительным зарядом на амино-конце спирали. Наиболее распространённое расположение α-спиралей находится на поверхности белков, где они обеспечивают взаимодействие с водной средой .
Внутренняя сторона спирали обычно содержит гидрофобные аминокислоты, а внешняя сторона гидрофильные аминокислоты. Таким образом, каждая третья из четырёх аминокислот в цепи будет гидрофобной, и поэтому эту аминокислоту можно легко обнаружить. В лейциновой молнии повторяющийся узор остатков лейцина на внешних сторонах двух соседних спиралей является, в значительной степени, характеризующем для данной структуры. Другие α-спирали, находящиеся в гидрофобном ядре белка или же в трансмембранных доменах белков , имеют более высокий процент гидрофобных аминокислот, которые равномернее распределены по цепи, что также служит хорошим маркером для данных частей белков. Качественное содержание аминокислот может быть хорошим маркером α-спиральной области. Регионы, имеющую большую концентрацию различных аминокислот , таких как аланин (A), глутаминовая кислота (E), лейцин (L) и метионин (M), а также более бедные по концентрации пролина (P), глицина (G), тирозина (Y) и серина (S), как правило, образуют α-спираль .
β-листы образованы Н-связями между в среднем 5—10 последовательными аминокислотами в одной части цепи и ещё 5—10 дальше по цепочке. Каждая цепь может проходить в одном и том же направлении, образуя параллельный лист, если цепи идут в разных направлениях, то образуется антипараллельный лист. Характер Н-связи различен в параллельной и антипараллельной конфигурации. Углы ψ и φ аминокислот в листах значительно варьируются в одной области Карты Рамачандрана . Предсказать местоположение β-листов в структуре белка сложнее, чем α-спиралей .
Петли представляют собой области белковой цепи, которые между α-спиралями и β-листами, различной длины и трёхмерной конфигурации и могут располагать, как на поверхности белка, так и ближе к ядру .
Петли шпилек, которые представляют собой полный оборот в полипептидной цепи, соединяющей две антипараллельные β-цепи, могут быть длиной до двух аминокислот. Петли могут взаимодействуют с окружающей средой(вода и другие растворители) и другими белками. Поскольку геометрия аминокислот в петлях не ограничена в пространством, как аминокислоты в области ядра, где очень плотная укладка цепи, и не так сильно влияют на правильную укладку белка, то там может происходить большее количество замен, вставок и делеций , которые не повлияют на функции белка. Таким образом, при выравнивании последовательностей, наличие этих мутаций (вставок, делеций, замен) может указывать на петлю. Позиции интронов в геномной ДНК иногда соответствуют местам петель в кодируемом белке, петли также имеют тенденцию иметь заряженные и полярные аминокислоты и часто являются компонентом сайтов связывания .

Третичная структура — пространственное строение (включая конформацию ) всех элементов вторичной структуры, состоящей из единственной цепи аминокислот. Спирализация линейной полипептидной цепи уменьшает её размеры примерно в 4 раза; а укладка в третичную структуру делает её в десятки раз более компактной, чем исходная цепь .
Поскольку ни полипептидная цепь, ни α-спирали и β-листы не дают представления об объёме , форме полипептидной цепи, перед исследователем всегда стоит необходимость определения трёхмерной или пространственной конфигурации белка.

Четвертичная структура — способ укладки в пространстве отдельных полипептидных цепей, обладающих одинаковой (или разной) первичной , вторичной или третичной структурой, и формирование единого в структурном и функциональном отношениях макромолекулярного образования. Специфичность четвертичной структуры белков проявляется в определённой конформационной автономии полипептидных фрагментов, входящих в состав макромолекулы белка. Вклад гидрофобных взаимодействий в стабилизацию третичной и четвертичной структуры белков весьма значителен: в случае третичной структуры на их долю приходится больше половины стабилизирующей силы.
Многие белки представляют собой сборки из нескольких полипептидных цепей. Примеры белков с четвертичной структурой включают гемоглобин , ДНК-полимеразу и различные ионные каналы
Алгоритмы предсказания вторичной структуры — это набор методов предсказания локальной вторичной структуры белков, основанных только на знании об их аминокислотной последовательности . Для белков предсказание состоит в соотнесении отдельных участков аминокислотной последовательности с наиболее вероятными классами вторичных структур, таких, как α-спирали, β-тяжи или петли . Точность предсказания определяется, как соотношение количества аминокислот, для которых предсказанный структурный класс совпал со структурным классом, определённым для этой аминокислоты [en] (или похожим алгоритмом, к примеру, ), к общему числу аминокислот в последовательности. Эти алгоритмы производят разметку аминокислотной последовательности белка в соответствии с принадлежностью аминокислот к одному из классов вторичной структуры, различающихся специфическими паттернами водородных связей и наборами двугранных углов. Для DSSP это 8 класcов, которые можно объединить в три группы: 3 класса спиралей (α-спираль, π-спираль и 3 10 -спираль), два класса β-структур (изолированные β-мостики и β-листы) и три вида петли (повороты, изгибы и неклассифицированные элементы, отвечающие характеристикам петли) . Чаще всего для оценки качества структуры используют упрощенную классификацию, в которой классы внутри этих трёх групп считаются тождественными . Алгоритмы предсказания вторичной структуры белка можно условно разделить на группы, основываясь на принципах, лежащих в их основе. Эти группы включают в себя статистические методы, методы ближайших соседей, методы, использующие нейронные сети, методы опорных векторов и методы, основанные на скрытых марковских моделях .
Ниже рассмотрены некоторые из этих алгоритмов.
Статистический основан на расчёте оценки вероятности принадлежности определённой аминокислоты к определённому классу вторичной структуры в базах данных. Предсказание делается относительно трёх классов вторичных структур: петли, β-листа и поворота. Цель алгоритма — найти участок из определённого для каждого класса вторичной структуры количества идущих подряд аминокислот, для каждой из которых оценка вероятности принадлежности к этому классу вторичной структуры больше заданного значения. На выход такие алгоритмы выдают предсказанные таким образом участки для каждого из трёх основных классов вторичных структур, картированные на последовательность.
Первый этап метода ближайших соседей ( ) заключается в поиске гомологичной последовательности, для которой известна трёхмерная структура. Учитывая локальные структурные особенности определённого аминокислотного остатка в трёхмерной структуре гомологичной последовательности, такие, как доступность для растворителя, полярность и вторичная структура, каждому аминокислотному остатку присваивается «класс окружения». Оценка вероятности принадлежности аминокислоты в центре исследуемого сегмента длиной n аминокислот к определённому классу вторичной структуры рассчитывается как логарифм частоты нахождения этой аминокислоты в окружении, к которому относится большинство её соседей, в базах данных.
Один из алгоритмов, использующих нейронные сети, , включает в себя четыре основных этапа: генерация позиционной весовой матрицы с помощью PSI-BLAST , первичное предсказание вторичной структуры и дальнейшая фильтрация предсказаний. Второй и третий этапы задействуют две нейросети. Для определения принадлежности аминокислоты к определённому классу вторичной структуры на вход первой нейронной сети подаётся фрагмент позиционной весовой матрицы размером 33x21, соответствующий фрагменту исходной последовательности в 33 аминокислоты с аминокислотой интереса по центру . Эта сеть имеет два скрытых слоя и три выходных узла, соответствующих трём предсказываемым классам вторичной структуры. Вторая нейронная сеть используется для фильтрации предсказаний первой сети и также обладает тремя выходного узлами для каждого класса вторичной структуры в центральной позиции исследуемого окна. На выход алгоритм выдаёт разметку аминокислотной последовательности по элементам вторичной структуры.
Помимо вышеописанного, классические алгоритмы с использованием скрытых марковских моделей, такие как алгоритм прямого-обратного хода , алгоритм Витерби и алгоритм Баума-Велша , могут быть оптимизированы для соотнесения аминокислотной последовательности с классами вторичных структур.
Наилучшие современные методы определения вторичной структуры белка достигают около 80 % точности . Точность ныне существующих методов предсказания вторичных структур оценивается такими еженедельно обновляющимися ресурсами, как от 12 апреля 2020 на Wayback Machine и от 24 февраля 2020 на Wayback Machine .
Большинство методов моделирования третичной структуры оптимизированы для моделирования третичной структуры отдельных белковых доменов. Этап, называемый анализом домена или предсказанием границы домена, обычно выполняется первым, чтобы разделить белок на потенциальные структурные домены. Как и в случае с остальными этапами предсказания третичной структуры, это можно сделать с помощью сравнения с известными структурами или ab initio только с помощью последовательности (обычно с помощью машинного обучения , задействующего ковариацию ) . Структуры отдельных доменов объединяются в одну окончательную третичную структуру в процессе, называемом сборкой доменов .
Методы моделирования ab initio- стремятся создавать трёхмерные белковые модели «с нуля», то есть они основаны на физических принципах, а не непосредственно на экспериментально полученных данных о структурах. Существует множество возможных подходов, которые либо пытаются имитировать сворачивание белка , либо применяют стохастические методы для поиска возможных решений (то есть поиск глобального максимума некой энергетической функции ) . Эти подходы, как правило, требуют огромных вычислительных ресурсов и, таким образом, могут быть применены только для крошечных белков. Для прогнозирования структуры белка ab initio для более крупных белков требуются более совершённые алгоритмы и большие вычислительные ресурсы, представленные либо мощными суперкомпьютерами (такими как Blue Gene или MDGRAPE-3 ), либо распределёнными вычислениями (такими как Folding@home , и Rosetta@Home ) .
Поскольку секвенирование стало более распространённым явлением в 1990-х годах, несколько групп исследователей использовали выравнивание белковых последовательностей для предсказания коррелированных мутаций, и была надежда, что эти совместно эволюционирующие остатки могут быть использованы для предсказания третичной структуры . Предполагается, что когда мутация одного аминокислотного остатка не является летальной, может возникнуть компенсаторная мутация для стабилизации взаимодействий между остатками. В ранних работах использовались так называемые локальные методы для расчёта коррелированных мутаций белковых последовательностей, при этом из-за независимого рассмотрения каждой пары остатков возникали ложные корреляции .
В 2011 году другой статистический подход продемонстрировал, что предсказанных коэволюционирующих остатков достаточно, чтобы предсказать трёхмерную укладку белка, при условии, что имеется достаточно последовательностей (необходимо > 1000 гомологичных последовательностей) . не использует моделирование по гомологии и может быть запущен на стандартном персональном компьютере даже для белков , состоящих из сотен остатков. Точность предсказаний этого и связанных с ним подходов, была продемонстрирована на многих структурах и .
Сравнительное моделирование структуры белка использует структуры, полученные ранее с помощью экспериментальных методов в качестве отправных точек. Это эффективно, так как, судя по всему, хотя число существующих белков огромно, количество третичных структурных мотивов , к которым принадлежит большинство белков , ограничено .
Эти методы также можно разделить на две группы :
Точное предсказание расположения боковых аминокислотных радикалов в структуре представляет собой отдельную проблему в прогнозировании структуры белка. Методы, которые решают проблему прогнозирования геометрии боковых радикалов, включают в себя и методы самосогласованного поля . Конформации боковых радикалов с низкой энергией обычно определяются на жёстком полипептидном остове и используют набор дискретных конформаций боковой цепи, «ротамеров». Принцип работы таких методов заключается в поиске набора ротамеров, минимизирующего общую энергию модели .
Эти методы используют библиотеки ротамеров, которые представляют собой наборы благоприятных конформаций для каждого типа остатка в белке. Библиотеки ротамеров могут содержать информацию о конформации, её частоте и стандартных отклонениях относительно средних значений торсионных углов, которые могут быть использованы при отборе вариантов . Библиотеки ротамеров получают с помощью структурной биоинформатики или другого статистического анализа конформаций боковых цепей в известных по экспериментальным данным структурах белков. Библиотеки ротамеров могут быть независимыми от остова , зависимыми от вторичной структуры или зависимыми от остова. Библиотеки ротамеров, не зависимые от остова, не используют информацию о конформации остова и рассчитываются по всем доступным боковым цепям определённого типа (например, первый пример библиотеки ротамеров, сделанный Пондером и Ричардсом в Йельском университете в 1987 году ). Библиотеки, зависящие от вторичной структуры , представляют собой различные торсионные углы и (или) частоты ротамеров для классов вторичных структур (альфа-спирали, бэта-листа или петли ). Зависящие от остова библиотеки ротамеров представляют собой конформации и (или) их частоты, зависящие от локальной конформации основной цепи, которая определяется торсионными углами фи и пси и не зависит от вторичной структуры . Современные версии этих библиотек, используемые в большинстве программ, представлены в виде многомерных распределений вероятности или частоты, где пики соответствуют конформациям торсионного угла, рассматриваемым как отдельные ротамеры.
Белок-белковый докинг (или Белок-белковое взаимодействие (ББВ) ) — метод молекулярного моделирования , позволяющий предсказать наиболее выгодную для образования устойчивого комплекса ориентацию и конформацию одной молекулы (лиганда) в центре связывания другой (рецептора). Данные о положении и конформации белков партнеров используются для предсказания силы взаимодействия посредством так называемых оценочных функций.
Так как до сих пор нет полных данных интерактома и не все белок-белковые взаимодействия обнаружены, при реконструкции сигнальных или метаболических карт взаимодействий используют различные вычислительные методы. Они позволяют устранить пробелы, предсказывая наличие тех или иных взаимодействий между узлами сети. С помощью вычислительных методов можно предсказать не только возможность ББВ, но также и их силу .
Ниже приведено несколько вычислительных подходов предсказания белок-белковых взаимодействий:

CASP (от англ. Critical Assessment of protein Structure Prediction — критическая оценка предсказания белковых структур) — масштабный эксперимент по предсказанию белковых структур. Проходит с 1994 года с периодичностью каждые два года . CASP объективно тестирует методы предсказания белковых структур и предоставляет независимую оценку структурного моделирования. Основная цель CASP — помощь в улучшении методов определения трёхмерной структуры белков из их аминокислотных последовательностей . Более 100 исследовательских групп принимают участие в проекте на постоянной основе. Один из главных принципов CASP — отсутствие у участников какой-либо предварительной информации о белке, кроме аминокислотной последовательности. По этой причине в CASP используется двойной слепой метод — ни организаторы, ни эксперты, ни участники не знают структуры тестируемых белков до окончания стадии предсказаний. Тестируемые белки — чаще всего ещё не разрешённые структуры, полученные методами рентгеноструктурного анализа и ЯМР .
Данное мероприятие помогает сравнить передовые методы предсказания структур белков и поиск «идеального» алгоритма, который лишь по аминокислотной последовательности сможет предсказывать третичную структуру белка .
На последнем CASP13 победила команда, которая использовала нейронную сеть AlphaFold . Так что, скорее всего, предсказание структур белков в будущем будут делать, используя именно нейронные сети .

Фолдит — онлайн-головоломка об укладке белка. Игра является частью исследовательского проекта и разработана в Вашингтонском университете . Предмет игры — наилучшим образом свернуть структуру выбранных белков; лучшие пользовательские решения анализируются учёными, которые могут с их помощью найти решение реальных научных проблем, связанных с поиском вакцин и биологическими инновациями. Большинство из лучших игроков «Фолдита» не имеют биохимического образования .
Целью данной игры является в предсказание трёхмерной структуры определённого белка с самым низким уровнем свободной энергии . Каждое задание публикуется на сайте на определённый срок, в течение которого пользователи соревнуются между собой.
Во время игры игроки интерактивно манипулируют молекулой , меняя углы остова белка и также расположение радикалов аминокислот. Игроки способны устанавливать ограничения на определённых участках («rubber bands») или «замораживать» их. Также пользователям предоставляется панель инструментов для выполнения автоматизированных задач, например, команда «wiggle» позволяет локально минимизировать энергию.
Пользователь получает информацию о том, насколько хорошо ему удаётся сворачивать белок, в форме баллов, которые начисляются, в частности, за образование новых водородных связей, сокрытие гидрофобных остатков внутрь молекулы и т. д. Также программа даёт игрокам подсказки, например подсвечивает участки, в которых определённые группы перекрываются и их следует развести, открытые гидрофобные участки, которые следует скрыть от воздействия воды и т. д. Сайт позволяет пользователям делиться друг с другом вариантами решений, обсуждать их .
Одним из первых алгоритмов предсказания вторичной структуры белка был метод ( англ. Chou–Fasman method ), опирающийся в первую очередь на вероятностные параметры, определённые с помощью относительных частот возникновения каждой аминокислоты в каждом типе вторичных структур . Точность метода Чоу-Фасмана составляет около 50—60 % .
Следующей примечательной программой был , названный по первым буквам фамилий его разработчиков, — метод, основанный на теории информации . Он использует вероятностный метод байесовского вывода . Метод GOR учитывает не только вероятность того, что аминокислота определённого типа включена в определённую вторичную структуру, но и условную вероятность того, что аминокислота включена в эту вторичную структуру с учётом вклада её соседей (при этом не предполагается, что соседи имеют такую же структуру) . Первоначальный метод GOR обладал точностью около 65 % и был значительно более успешен в предсказании альфа-спиралей , нежели чем бета-листов , которые он часто неверно предсказывал как или неорганизованные участки .
Ещё одним большим шагом вперёд стало использование методов машинного обучения : первые методы нейронных сетей были использованы в программах для предсказания вторичных структур белков. В качестве обучающих выборок они использовали последовательности белков с экспериментально полученными структурами для определения общих мотивов , связанных с определённым расположением вторичных структур . Эти методы более чем на 70 % точны в своих предсказаниях, хотя количество бета-тяжей так же часто занижается из-за отсутствия информации о трёхмерной структуре, которая позволила бы оценить паттерны водородных связей, которые могут способствовать формированию бета-листа . от 21 июля 2011 на Wayback Machine и от 7 апреля 2020 на Wayback Machine являются одними из самых известных программ для предсказания вторичной структуры белка, основанных на нейронных сетях . Позже метод опорных векторов оказался особенно полезным для предсказания поворотов, которые трудно идентифицировать статистическими методами .
Расширения методов машинного обучения используются для предсказания более точных локальных свойств белков, таких как торсионные углы остова в областях с неклассифицированной структурой. И метод опорных векторов, и нейронные сети были использованы для решения этой проблемы . Совсем недавно программа от 12 апреля 2020 на Wayback Machine позволила точно прогнозировать реальные торсионные углы и успешно использовать эту информацию для прогнозирования структуры ab initio .