Молекулярная биология
- 1 year ago
- 0
- 0

Моти́в в молекулярной биологии — относительно короткая последовательность нуклеотидов или аминокислот , слабо меняющаяся в процессе эволюции и, по крайней мере предположительно, имеющая определённую биологическую функцию . Под мотивом иногда подразумевают не конкретную последовательность, а каким-либо образом описанный спектр последовательностей, каждая из которых способна выполнять определённую биологическую функцию данного мотива .
Мотивы встречаются в живых организмах повсеместно и выполняют множество жизненно важных функций, таких как регуляция транскрипции и трансляции (в случае нуклеотидных мотивов), посттрансляционная модификация и клеточная локализация белков, и частично обуславливают их функциональные свойства ( лейциновая молния ) . Они широко используются в биоинформатике для предсказания функций генов и белков, построения карт регуляции, важны для многих задач генной инженерии и молекулярной биологии в целом .
В связи с практической важностью мотивов, разработаны как биоинформатические методы их поиска ( MEME , Gibbs Sampler), так и методы поиска мотивов in vivo ( ChIP-seq , ChIP-exo). Последние довольно часто дают приблизительные координаты мотивов и их результаты затем уточняются биоинформатическими методами .Для удобства хранения мотивов в базах данных используются их разные, отличающееся степенью детальности, представления, наиболее распространенными из которых являются консенсус и позиционная весовая матрица .
Следует отличать мотив от консервативных участков в близкородственных организмах, необладающих значимыми биологическими функциями, где мутационный процесс не успел ещё достаточно их изменить .
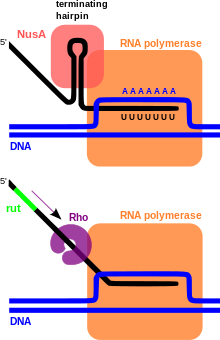
В случае с ДНК чаще всего мотивы представляют собой короткие последовательности, являющиеся сайтами связывания для белков, таких, как нуклеазы и транскрипционные факторы , или вовлечённые в важные регуляторные процессы уже на уровне РНК , такие как посадка рибосомы , процессинг мРНК и терминация транскрипции .
Изучение мотивов в ДНК стало возможным благодаря появлению в 1973 году процедуры секвенирования ДНК (определения последовательности нуклеотидов фрагмента ДНК). Первыми были определены последовательности lac -оператора и лямбда-оператора . Однако до появления более производительных методов секвенирования , количество последовательностей мотивов оставалось достаточно малым. К концу 1970-х годов появилось множество примеров мутантных последовательностей (сайтов), связывающих транскрипционные факторы и последовательностей с изменённой специфичностью . С увеличением количества последовательностей, стали развиваться и методы теоретического предсказания мотивов. В 1982 году была впервые сконструирована позиционно-весовая матрица (ПВМ) мотива сайта инициации трансляции. С помощью построенной ПВМ были предсказаны другие сайты инициации трансляции . Этот подход оказался достаточно мощным и до сих пор в разных формах применяется для поиска известных мотивов в геномах, а конкретные методы различаются только видом весовой функции . Однако подход, основанный на построении ПВМ на базе уже имеющихся последовательностей, не позволял находить принципиально новые мотивы, что является более сложной задачей. Первый алгоритм, решавший эту задачу, был предложен Галласом с коллегами в 1985 году . Этот алгоритм был основан на поиске общих слов в наборе последовательностей и давал большой процент ложноотрицательных результатов, однако он стал основой для целого семейства алгоритмов . Позднее были разработаны более точные вероятностные методы: алгоритм MEME , основанный на процедуре максимизации ожидания и алгоритм , также основанный на процедуре максимизации ожидания . Оба метода оказались очень чувствительными и используются в настоящее время для предсказания мотивов в наборах последовательностей.
После разработки мощных средств для предсказания мотивов связывания транскрипционных факторов и установления соответствия между достаточным количеством транскрипционных факторов и мотивов, стало возможным предсказывать функции оперона, лежащего поблизости от мотива по специфичности транскрипционного фактора, с ним связывающегося и наоборот, предсказывать транскрипционный фактор по генам в опероне, лежащем рядом с определённым мотивом .
Характерными примерами регуляции транскрипции, осуществляемой с помощью белка, распознающего специальный мотив, являются:
Одними из наиболее известных примеров регуляции трансляции при помощи мотив-распознающих регуляторов являются:
Сила взаимодействия белка или РНК с ДНК мотивом зависит в первую очередь от последовательности данного мотива. Различают «сильные» мотивы, дающие сильное взаимодействие с белком или РНК и «слабые» мотивы, с которыми взаимодействие слабее. Практически всегда удаётся получить так называемую «консенсусную последовательность» («консенсус»), то есть такую последовательность, в каждой позиции которой стоит буква, наиболее часто встречающаяся в соответствующей позиции в последовательностях мотивов из разных организмов. Консенсусная последовательность принимается за самую сильную, каковой она почти всегда и является . Более слабые мотивы получаются из неё с помощью небольшого (чаще всего 1—3) числа замен .
В процессе эволюции сила мотивов регулируется с помощью естественного отбора, причём мотив может становиться как сильнее, так и слабее . Характерным примером такой подстройки силы мотива может служить изменчивость последовательности Шайна — Дальгарно (ШД). Есть тесная корреляция между необходимым организму количеством транслируемого белка и силой ШД перед ним .
Важно отметить, что в случае с ШД, хотя сила связывания белка и напрямую коррелирует с силой связывания 16S-субъединицы рибосомы , в связи с особенностями инициации трансляции, консенсусная последовательность не обязательно будет гарантировать наиболее эффективную трансляцию (из-за затруднённого ухода рибосомы с сайта инициации ) . Поэтому последовательность Шайна — Дальгарно чаще всего содержит 4—5 нуклеотидов из консенсусной последовательности при длине последней примерно в 7 нуклеотидов .
Не всегда наличие мотива, явно выполняющего биологически значимую роль, влечёт за собой наличие белка-регулятора. Регуляция также может осуществляться за счёт связывания РНК с каким-либо низкомолекулярным веществом . На этом принципе построены — структуры, образующиеся на РНК во время транскрипции, способные связывать малые молекулы . Связывание молекулы влияет на способность рибопереключателя останавливать транскрипцию или препятствовать трансляции. В этом случае важной оказывается не последовательность нуклеотидов как таковая, а наличие комплементарных нуклеотидов на нужных местах в последовательности .
Регуляция трансляции также может осуществляться только за счёт образуемой нуклеиновой кислотой вторичной структуры.
Зачастую, мотивы, связывающие транскрипционные факторы, имеют вид прямых повторов некоторой последовательности, обратных повторов или палиндромных последовательностей . Это можно объяснить работой транскрипционных факторов в виде димеров белков, в которых каждый из мономеров связывает одну и ту же последовательность. Встречаются также мотивы большей повторности . Такое строение мотивов обеспечивает большую резкость реакции на изменение внешних условий. К примеру, если связывание зависит от концентрации одного вещества в клетке, то получаем зависимость силы реакции клетки, описываемую уравнением Михаэлиса — Ментен . С увеличением числа связывающихся единиц белка (будем считать, что действие связывания белка с мотивом проявляется только в случае связывания со всеми повторами) зависимость всё больше становится похожей на сигмоиду , в пределе стремясь к функции Хевисайда , описывающей один из главных принципов реагирования живых систем на многие воздействия — закон «всё или ничего» ( англ. all-or-nothing law ) , к примеру, формирования потенциала действия .
Для белков следует различать
Мотивы в первичной структуре похожи на мотивы в нуклеиновых кислотах. Характерными примерами таковых являются:
В белках структурные мотивы описывают связи между элементами вторичной структуры. Такие мотивы часто имеют участки переменной длины, которые в некоторых случаях могут и вовсе отсутствовать .
Кроме бета-шпильки выделяют и множество других мотивов, функция которых состоит в формировании структурного каркаса белка .
Близким к термину структурный мотив белка является — характерное расположение элементов вторичной структуры. В силу своей схожести термины часто используются один вместо другого и грань между ними размыта .
Изначально имеется набор мотивов из разных последовательностей и ставится задача :
Существует несколько общепризнанных способов представления мотивов . Часть из них подходит как для белков, так и для нуклеотидов, другая часть — только для белков или нуклеотидов.
Строгим консенсусом мотива назовем строчку, состоящую из самых представленных букв в множестве реализаций мотива. На практике, указывается не просто наиболее частая буква в данной позиции, но и, если максимальная частота встречаемости какой-либо буквы в данной позиции меньше заданного порога, то на этом месте в консенсусе ставится
x
(любая буква алфавита). По такому консенсусу мы почти наверняка находим последовательности, реально являющиеся мотивами, но упускаем большое число мотивов, отличающихся от консенсуса на несколько замен
. Ниже приведён пример строгого консенсуса для участка мотива пяти взятых из
UniProt
белков с мотивом лейциновой молнии (порог был взят равным 80 %):
| Номер позиции | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| UniProt ID | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 |
| O35048 | L | S | P | C | G | L | R | L | I | G | A | H | P | I | L |
| Q6XXX9 | L | G | Q | D | I | C | D | L | F | I | A | L | D | V | L |
| Q9N298 | L | G | Q | V | T | C | D | L | F | I | A | L | D | V | L |
| Q61247 | L | S | P | L | S | V | A | L | A | L | S | H | L | A | L |
| B0BC06 | L | T | I | G | Q | Y | S | L | Y | A | I | D | G | T | L |
| Консенсус | L | x | x | x | x | x | x | L | x | x | x | x | x | x | L |
Нестрогим консенсусом назовем последовательность списков букв, наиболее представленных на соответствующем месте. Описываются все или наиболее часто встречающиеся буквы в данной позиции (обычно устанавливается минимальный порог частоты) . Фактически, мотив описывается при помощи регулярного выражения . В качестве обозначений используют:
ABC
— строка из символов алфавита, обозначающая последовательность символов, следующих друг за другом;
[ABC]
— любая строка символов, взятых из алфавита в квадратных скобках соответствует любому из соответствующих символов; например [ABC] соответствует или A или B или C;
{ABC..DE}
— любая строка символов, взятых из алфавита, соответствует любой аминокислоте, кроме тех, что находятся в фигурных скобках; например
{ABC}
соответствует любой аминокислоте, кроме
A
,
B
и
C
;
x
в нижнем регистре — любой символ алфавита.
В случае с таким представлением приходится балансировать между чувствительностью консенсуса (количеством реальных мотивов, которые им получится отыскать) и специфичностью (способностью метода отбраковывать мусорные последовательности)
. Ниже приведен пример нестрого консенсуса для тех же пяти последовательностей белков, что и для строго консенсуса (порог был взят равным 20 %). Видим, что в позиции 10 мотив не совсем объективен —
лейцин
(
L
) и
изолейцин
(
I
) — очень близкие по свойствам аминокислоты, и логично было бы их обе занести в консенсус.
| Номер позиции | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| UniProt ID | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 |
| O35048 | L | S | P | C | G | L | R | L | I | G | A | H | P | I | L |
| Q6XXX9 | L | G | Q | D | I | C | D | L | F | I | A | L | D | V | L |
| Q9N298 | L | G | Q | V | T | C | D | L | F | I | A | L | D | V | L |
| Q61247 | L | S | P | L | S | V | A | L | A | L | S | H | L | A | L |
| B0BC06 | L | T | I | G | Q | Y | S | L | Y | A | I | D | G | T | L |
| Консенсус | L | [SG] | [PQ] | x | x | C | D | L | F | I | A | [LH] | D | V | L |
PROSITE использует ИЮПАК для обозначения однобуквенных кодов аминокислот, за исключением символа конкатенации «-», используемого между элементами паттерна. При использовании PROSITE добавляется несколько символов, облегчающих представление белкового мотива :
<
' — шаблон ограничивается N-концом последовательности;
>
' — шаблон ограничивается C-концом последовательности;
Если
e
— шаблон элемента, и
m
и
n
два десятичных целых числа и
m
<=
n
, то:
e(m)
эквивалентно повторению
e
ровно
m
раз;
e(m,n)
эквивалентно повторению
e
ровно
k
раз для любого целого
k
удовлетворяющего условию:
m
<=
k
<=
n
;
Пример:
мотив домена с сигнатурой C2H2-type
цинкового пальца
выглядит следующим образом:
C-x(2,4)-C-x(3)-[LIVMFYWC]-x(8)-H-x(3,5)-H
Позиционной весовой матрицей называется такая матрица, столбцы которой соответствуют позиции в последовательности, а строчки соответствуют буквам в алфавите. Значениями этой матрицы являются частоты (или монотонные функции от частот) встречаемости данной буквы в данной позиции на последовательности. При этом обычно, чтобы исключить нулевые частоты к числу встреч каждой буквы позиции добавляют некоторое число, исходя из априорного распределения букв в подобных последовательностях (к примеру, вводят поправку Лапласа ). Данный подход, как и предыдущие, неявно предполагает, что позиции в мотиве независимы, чего на самом деле не наблюдается даже для нуклеотидных последовательностей .
Пусть у нас есть 7 последовательностей ДНК, представляющих собой мотив :
| Номер позиции | ||||||||
|---|---|---|---|---|---|---|---|---|
|
Номер
последовательности |
1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| 1 | A | T | C | C | A | G | C | T |
| 2 | G | G | G | C | A | A | C | T |
| 3 | A | T | G | G | A | T | C | T |
| 4 | A | A | G | C | A | A | C | C |
| 5 | T | T | G | G | A | A | C | T |
| 6 | A | T | G | C | C | A | T | T |
| 7 | A | T | G | G | C | A | C | T |
Позиционная матрица для них будет иметь следующий вид (
+1
— учёт правила Лапласа)
:
| Номер позиции | ||||||||
|---|---|---|---|---|---|---|---|---|
| Нуклеотид | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| A | 5 + 1 | 1 + 1 | 0 + 1 | 0 + 1 | 5 + 1 | 5 + 1 | 0 + 1 | 0 + 1 |
| C | 1 + 1 | 0 + 1 | 1 + 1 | 4 + 1 | 2 + 1 | 0 + 1 | 6 + 1 | 1 + 1 |
| G | 0 + 1 | 1 + 1 | 6 + 1 | 3 + 1 | 0 + 1 | 1 + 1 | 0 + 1 | 0 + 1 |
| T | 1 + 1 | 5 + 1 | 0 + 1 | 0 + 1 | 0 + 1 | 1 + 1 | 1 + 1 | 6 + 1 |
Частоты можно пронормировать на общее число последовательность, тем самым получив оценку вероятности встречи данного нуклеотида в данной последовательности (собственно, обычно в таком представлении и хранится PWM) :
| Номер позиции | ||||||||
|---|---|---|---|---|---|---|---|---|
| Нуклеотид | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| A | 0,55 | 0,18 | 0,09 | 0,09 | 0,55 | 0,55 | 0,09 | 0,09 |
| C | 0,18 | 0,09 | 0,18 | 0,45 | 0,27 | 0,09 | 0,64 | 0,18 |
| G | 0,09 | 0,18 | 0,64 | 0,36 | 0,09 | 0,18 | 0,09 | 0,09 |
| T | 0,18 | 0,55 | 0,09 | 0,09 | 0,09 | 0,18 | 0,18 | 0,64 |

Для большей точности можно учитывать зависимость соседних позиций в мотиве с помощью скрытых марковских моделей первого и более высоких порядков . Этот подход сопряжён с некоторыми трудностями, так как для его применения необходимо наличие достаточно представительной выборки вариантов мотивов. В случае предыдущего примера имеем:

В случае мотивов, содержащих участки переменного размера и нуклеотидного состава, можно было бы вводить отдельно модель для этих участков, отдельно — для консервативных, а затем «склеивать» их в одну модель путём добавления промежуточных «молчащих» состояний и вероятностей перехода в них и из них .
В случае мотивов, формирующих вторичные структуры (РНК-переключатели) в РНК, в элементах вторичной структуры важно учитывать возможность спаривания нуклеотидов . С этой задачей справляются СКС . Однако обучение СКС требует ещё большего размера выборки, чем HMM, и сопряжено с рядом трудностей .
В тех случаях, когда важна скорость поиска и допустим пропуск некоторых вхождений нашего мотива, исследователи прибегают к различным уловкам, позволяющим с приемлемой точностью зашифровать пространственную структур биополимера (РНК или белка) путём расширения алфавита .
Оперон Escherichia coli репрессор лактозы LacI ( PDB chain A) и ген активатор катаболизма ( PDB chain A) оба имеют мотив спираль-поворот-спираль, но их аминокислотные последовательности не очень схожи. Группой исследователей был разработан код, который они назвали «трёхмерный код цепи», представляющий структуру белка в виде строки из писем. Эта схема кодирования, по мнению авторов, показывает сходство между белками гораздо более отчётливо, чем аминокислотные последовательности :
Пример : сравнение двух упомянутых выше белков при помощи этой схемы кодирования :
| PDB ID | 3D-code | Amino acid sequence |
|---|---|---|
1lccA
|
TWWWWWWWKCLKWWWWWWG
|
LYDVAEYAGVSYQTVSRVV
|
3gapA
|
KWWWWWWGKCFKWWWWWWW
|
RQEIGQIVGCSRETVGRIL
|
| Сравнение | Видно явное сходство между белками | По аминокислотной последовательности белки сильно отличаются |
где
W
соответствует α-спирали, и
E
и
D
соответствует β-нити.
В данной работе с целью применения алгоритма поиска, схожего с BLAST , нуклеотидный алфавит (ATGC, так как поиск осуществлялся в геноме) был расширен за счёт комбинирования нуклеотидов и трех символов, характеризующих их предположительное направление спаривания :
(
— нуклеотид спарен с нуклеотидом справа;
)
— нуклеотид спарен с нуклеотидом слева;
.
— нуклеотид не спарен.
Таким образом получалось 12 букв нового алфавита (4 нуклеотида * 3 «направления»), при правильном использовании позволяющий осуществлять BLAST-подобный поиск, названный авторами foldedBlast .

Для визуального представления мотивов часто используют логотип последовательностей — графического представления консервативности каждой позиции в мотиве. При этом данную визуализацию можно успешно применять как и в случае представления мотива в виде консенсуса или позиционной весовой матрицы , так и для представления HMM модели последовательности, как это сделано в базе белковых семейств Pfam .
Кроме того, если использовать, к примеру, яркость каждой нуклеотида в мотиве как индикатор того, насколько часто ему соответствует в этом же мотиве комплементарный нуклеотид, то можно частично представлять и информацию о вторичной структуре мотива. Так сделано, например, в биоинформатическом веб-сервисе .
В случае поиска в нуклеотидных последовательностях мотивов, отвечающих за связывание регуляторных белков пользуются соображением, что они [мотивы] меняются сравнительно медленно, а значит, если взять организмы, достаточно далёкие друг от друга, чтобы в высоковариабельных позициях их последовательностей успели накопиться мутации, а сайты измениться сильно ещё не успели, то можно пользоваться правилом «что консервативно — то важно» . После получения последовательностей, в которых предполагается наличия специфичного мотива, в основном используют два подхода к поиску последовательности мотива — филогенетический футпринтинг и сведение задачи к задаче поиска вставленного мотива .
Филогенетический футпринтинг — полуавтоматический метод. Последовательности обрабатываются программой множественного выравнивания , и в получившемся выравнивании исследователем ищутся паттерны, которые можно считать мотивами. Одним из наиболее успешных примеров применения данного подхода можно считать расшифровку способа кодирования нерибосомных пептидов нерибосомными пептид-синтетазами (NRPS) . Данный метод не позволяет полностью автоматизировать процесс поиска мотивов, но при этом и не имеет столь сильных ограничений, как следующие.
В случае с мотивами без (почти без) разрывов и без (почти без) участков переменной длины возможно свести задачу поиска мотива к задаче поиска вставленного мотива ( англ. Planted motif search ) .
Формулировка задачи следующая: « На вход предоставлены n строк s 1 , s 2 , …, s n длины m, каждая составленная из символов алфавита A, и два числа — l и d. Найдите все строки x длины l такие, что любая из предоставленных строки содержит хотя бы одну подпоследовательность, находящуюся от x на расстоянии Хэмминга не больше d » .
Так как в общем случае неизвестно, все ли полученные нами последовательности имеют искомый мотив, а также неизвестна его точная длина, то обычно задачу решают эвристическими методами — максимизируя вероятность найденного мотива при данных последовательностях. На этом принципе построены программы MEME и GibbsSampler .
Если задать минимальный порог на число последовательностей, в которых должен содержаться мотив, и как-либо ограничить его длину, то можно использовать и точные способы решения данной задачи, к примеру — алгоритм RISOTTO . Некоторые из них позволяют снимать часть ограничений на искомый мотив — в RISOTTO искомый мотив может иметь разрывы, состоять из нескольких частей.
Однако эти методы редко дают результаты лучше, чем MEME и GibbsSamler, а работают они значительно дольше .
Метод анализа ДНК-белковых взаимодействий, комбинирующий идеи иммунопреципитации хроматина (ChIP) и высокоэффективном секвенировании ДНК (белок пришивается к ДНК, затем кусочки ДНК, пришившиеся к белку отправляются на секвенирование). В ходе работы метода получаются участки длиной около 150 нуклеотидов, которые затем можно анализировать in silico на наличие мотива .
Как и в случае использования метода ChIP-seq проводится иммунопреципитации хроматина (ChIP), затем сшивка с белком обращается и полученная ДНК гибридизуется с ДНК-микрочипом . Метод ChIP-on-chip дешевле, чем ChIP-seq, однако сильно уступает последнему в точности .
Также метод, основанный на иммунопреципитации хроматина (ChIP). Использование экзонуклеазы фага λ , деградирующей ДНК только с 5'-конца и только в случае отсутствия контакта с белком, позволяет добиваться точности порядка нескольких нуклеотидов в определении положения сайта связывания белка .
Итеративный метод поиска нуклеотидных последовательностей, хорошо связывающихся с данным белком . Процедура в общем случае выглядит так:
Делается гибридный белок из изучаемого белка и адениновой ДНК- метилтрансферазы Dam . В естественных условиях аденин в большинстве эукариот не метилируется. Когда же гибридный белок связывается с каким-либо сайтом в ДНК организма, метилтрансферазная часть модифицирует аденины в районе этого сайта, что позволяет затем с помощью эндонуклеаз рестрикции выделить участок, на котором с большой долей вероятности находится искомый мотив.