Бензоиновая конденсация
- 1 year ago
- 0
- 0


Альдольная конденсация ( альдольно-кротоновая конденсация , альдольная реакция ) — химическая реакция между двумя молекулами альдегида или кетона в присутствии кислоты или основания с образованием альдоля (β-гидроксиальдегида или β-гидроксикетона), а в некоторых случаях — продукта дегидратации альдоля (α,β-ненасыщенного альдегида или кетона) .
Реакция была открыта независимо Шарлем Адольфом Вюрцем и Александром Бородиным в 1872 году, а в 1880 году Шмидт предложил разновидность альдольной конденсации — — и впервые провёл альдольную конденсацию в условиях основного катализа .

Альдольная реакция является одним из самых важных методов в органическом синтезе . Разработаны способы направленного проведения этой реакции, её региоселективные и стереоселективные аналоги. Реакция представляет большую ценность в синтезе природных соединений. Альдольная конденсация протекает также в биологических системах.
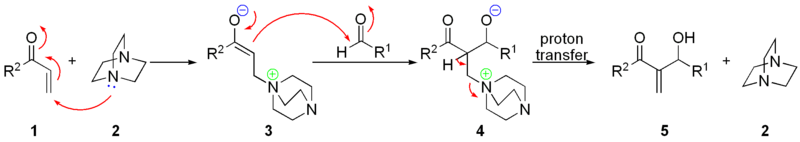
Иногда термин альдольная конденсация применяют к ряду реакций, имеющих подобный механизм, но другие реагенты и продукты ( реакции Клайзена , Кнёвенагеля , , Перкина , и Реформатского ) .
Для удобства описания реакций вещества, взаимодействующие в альдольной конденсации, разделяют в зависимости от их роли. Альдегид или кетон, вступающий в реакцию карбонильной группой, называют карбонильной компонентой , а вещество, участвующее в реакции α-метиленовой группой и превращающееся под действием катализатора в енолят-ион или енол, называют метиленовой компонентой . Очевидно, что карбонильной компонентой теоретически может быть любой альдегид или кетон, а метиленовой компонентой лишь тот, у которого есть хотя бы один α-водородный атом. Например, метиленовой компонентой не могут быть формальдегид , бензальдегид или .
Карбонильные компоненты различаются по реакционной способности, которая определяется величиной частичного положительного заряда на атоме углерода карбонильной группы. В связи с этим уменьшение активности карбонильных соединений наблюдается в следующем ряду: формальдегид — альдегиды — кетоны .
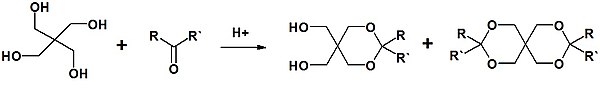
Формальдегид по реакционной способности превосходит все другие альдегиды, поэтому часто его используют как карбонильную компоненту в реакциях с другими альдегидами, не опасаясь, что последние будут конденсироваться сами с собой. Если формальдегид используется в избытке, то реакция не останавливается на стадии образования альдоля, а протекает дальше с участием других α-водородных атомов метиленовой компоненты. Данное явление используется в синтезе пентаэритрита , в основе которого лежит альдольная реакция между формальдегидом и ацетальдегидом .

В качестве метиленовых компонент в реакции могут выступать не только карбонильные соединения (альдегиды и кетоны), но и любые другие, обладающие CH-кислотностью, то есть те, от углеродного атома которых основанием можно отщепить протон (например, производные карбоновых кислот ) . Наличие акцепторных групп в структуре альдегида или кетона повышает его кислотность и облегчает отщепление протона из α-положения .
Если роль метиленовой компоненты выполняет несимметричный кетон, то отщепление α-метиленового протона может происходить от двух неодинаковых α-положений, что ведёт к образованию двух разных продуктов конденсации. Предпочтительным же является отщепление протона от более замещённого атома углерода, поскольку при этом образуется более устойчивый , в котором двойная связь содержит больше заместителей (см. правило Зайцева ) . Существуют, однако, методы получения енолятов с менее замещённой двойной связью, основанные на использовании пространственно затруднённых оснований. В этом случае направление реакции енолизации определяется не устойчивостью енолята, а пространственными эффектами [ источник не указан 3795 дней ] .
С учётом того, что в альдольной реакции принимает участие два карбонильных соединения, существует несколько принципиальных комбинаций реагентов, в случае которых реакция протекает различным способом .

Альдольная конденсация может проводиться в условиях кислотного или основного катализа (последний используется чаще) .
Реакция, катализируемая основаниями, включает в себя три стадии .

При высокой концентрации альдегида скорость реакции лимитирует стадия отщепления протона, однако при разбавлении реакция приобретает второй порядок по альдегиду. Для большинства же реакций с участием кетонов лимитирующей стадией является стадия присоединения енолята к карбонильному соединению .
Реакция может также катализироваться кислотами. В этом случае происходит активация карбонильной компоненты путём протонирования её карбонильной группы . Метиленовая компонента превращается в енол, который обладает нуклеофильной реакционной способностью (хотя и значительно меньшей, чем енолят-ион) и на следующей стадии присоединяется к активированной карбонильной компоненте . Скорость всего процесса определяется скоростью второй стадии .
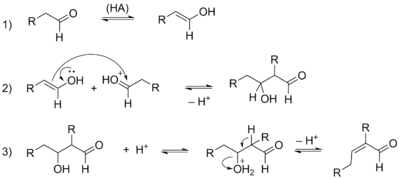
Как правило, наиболее распространёнными продуктами в кислых условиях являются α,β-непредельные соединения, поскольку дегидратация, следующая за образованием альдоля, протекает достаточно быстро .
Все стадии альдольной конденсации являются равновесными (включая стадию дегидратации ), поэтому её продукты при обработке щёлочью можно снова расщепить до исходных реагентов. Такой процесс известен как ретроальдольная реакция .
Классические условия проведения альдольной конденсации включают обработку карбонильного соединения щёлочью или другим основанием в водной или водно-спиртовой среде при 0—5 °С. В этих условиях образуется альдоль (сокр. от альдегид и алкоголь ) — β-гидроксиальдегид. В более жёстких условиях (например, при нагревании) альдоль теряет молекулу воды с образованием — α,β-ненасыщенного альдегида. При проведении реакции в кислой среде остановить реакцию на стадии образования альдоля сложно, и продуктом реакции оказывается α,β-ненасыщенное карбонильное соединение .
Многие альдольные конденсации не слишком чувствительны к концентрации катализатора, и в большинстве случаев достаточно небольшого количества основания, чтобы получить приемлемый выход. Избыток основания способствует ретроальдольной реакции и дегидратации. Кислотно-катализируемые реакции также малочувствительны к концентрации кислоты и, в основном, легко приводят к α,β-непредельным продуктам .
В качестве катализаторов было использовано множество веществ, но наиболее часто применяется лишь ограниченное количество. Гидроксиды щелочных и щёлочноземельных металлов нашли наиболее широкое применение. Часто используется гидроксид натрия , но гидроксид калия так же эффективен. Если альдегид чувствителен к щелочам, в качестве катализатора выбирают гидроксид кальция или гидроксид бария . В случаях, когда гидроксид натрия неэффективен, применяют алкоголяты щелочных металлов (чаще всего — этилат натрия в этаноле ). Определённую область применения нашли соли щелочных металлов и слабых кислот ( карбонат натрия , карбонат калия ), позволяющие поддерживать постоянное значение pH реакционной среды. Также в качестве катализаторов применяются ионообменные смолы , некоторые реактивы Гриньяра и т. д.
Первичные и вторичные амины являются ценными катализаторами в реакциях конденсации альдегидов, чувствительных к щелочам, а также карбонильных соединений с высоким содержанием енольной формы (например, 1,3-дикетонов). Наиболее эффективны в этом смысле пирролидин и пиперидин . Добавление уксусной кислоты к этим аминам ускоряет конденсацию (иногда используются непосредственно ацетаты этих аминов) .
Кислотные катализаторы применяются не так часто, поскольку они дают более низкие выходы, а процедура очистки продукта более сложна. В случае конденсации альдегидов с альдегидами образуются смолы. Основным катализатором из кислот является хлороводород , однако при его участии в качестве продуктов могут получаться β-галогенкарбонильные соединения. Также используются серная кислота , , реже — азотная кислота , трифторид бора , оксихлорид фосфора , уксусный ангидрид и другие кислоты .
Выбор растворителя зависит от растворимости реагентов. Наиболее распространены вода, этанол и водно-спиртовые смеси. Для реакционноспособных альдегидов хорошо подходят гетерогенные смеси (водный гидроксид натрия — диэтиловый эфир ). В случае использования сильных оснований требуются апротонные растворители , а хлороводород часто используют вовсе без растворителя .
Наиболее хорошие выходы достигаются при 5—25 °С. Скорость реакции при этом обычно достаточна для того, чтобы провести реакцию за 12 или 24 часа. Если в альдольной конденсации получаются неустойчивые продукты, температуру понижают до 0—5 °С. Реакции, катализируемые кислотами, также обычно требуют пониженной температуры .
Как правило, для проведения реакции берут стехиометрические количества двух карбонильных соединений. Важное исключение составляют реакции между активными альдегидами и кетонами, когда используется большой избыток кетона, чтобы предотвратить самоконденсацию альдегида. Самоконденсации альдегида можно также избежать путём медленного добавления альдегида к избытку кетона, содержащего катализатор .
Избыток альдегида может быть полезен в том случае, когда кетон недостаточно активен в альдольной реакции, а альдегид неспособен конденсироваться сам с собой (как, например, бензальдегид) .
Перекрёстная альдольная конденсация (конденсация двух разных карбонильных соединений) имеет ограниченную область применения. Это связано с рядом причин, в первую очередь, с образованием нескольких ожидаемых продуктов реакции, также образованием побочных продуктов, продуктов самоконденсации и поликонденсации. Также в случае использования несимметричных кетонов в реакции конденсации участвуют два изомерных енолят-иона, полученных при депротонировании альтернативных α-положений, что приводит к увеличению числа возможных продуктов. Использование протонного растворителя не благоприятствует образованию альдоля, а ведёт к образованию α,β-непредельного продукта. Кроме того реакция обратима и не может быть доведена до завершения, если продукт реакции неустойчив. В связи с этим в последнее время разработаны методы повышения региоселективности перекрёстной конденсации, связанные с использованием литиевых, борных, цинковых и других енолятов в апротонной среде. Суть данных подходов заключается в предварительном количественном превращении метиленовой компоненты в енолят с последующим добавлением в реакционную смесь второго карбонильного соединения, выполняющего роль карбонильной компоненты .
В данном подходе один из участников реакции нацело превращают в литиевый енолят под действием сильного основания (например, диизопропиламида лития LDA в тетрагидрофуране ) при −78 °C, а затем добавляют второй субстрат, являющийся карбонильной компонентой. При этом присоединение енолята к карбонильной группе происходит быстрее, чем перенос протона между компонентами или изомеризация енолята, поэтому образуется продукт, заданный порядком смешивания реагентов. Недостатком литиевых енолятов является их высокая основность, которая сужает круг используемых субстратов . Также данный подход редко применяется к альдегидам, поскольку для них реакция самоконденсации протекает слишком быстро, из-за чего не удаётся получить устойчивый литиевый енолят .

В 1973 году японский химик предложил силильные еноляты как альтернативу литиевым енолятам. В его варианте альдольная конденсация проводится между силильным енолятом как эквивалентом енола , а для активации карбонильной компоненты добавляется кислота Льюиса , например, трифторид бора или хлорид титана(IV) . Силиеноляты легко получить, они удобны в обращении. Основным варьируемым параметром в данной реакции является природа кислоты Льюиса: проводя реакцию с теми или иными солями металлов, регулируют стереохимию протекания реакции .

Реакция Мукаямы в данном виде является аналогом альдольной реакции, катализируемой кислотой. Эту реакцию также можно проводить другим способом, аналогичным основнокаталитической альдольной конденсации. В этом случае реакцию катализируют фторид-ионом F - (обычно используют фторид тетрабутиламмония или другие более сложные источники фторида), а силиеноляты выступают как эквиваленты енолят-ионов .
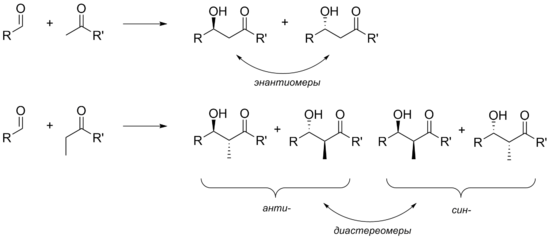
В некоторых случаях при протекании альдольной конденсации в продукте возникает новый и, следовательно, образуется смесь двух стереоизомеров . Такая ситуация наблюдается в тех случах, когда в реакцию с альдегидами вступают еноляты метилзамещённых кетонов (образуется пара энантиомеров ). В случае же енолятов этилкетонов в продукте возникает сразу два стереоцентра, поэтому конечный продукт представлен двумя диастереомерами с анти - и син -расположением заместителей, причём каждому из диастереомеров соответствует пара энантиомеров. Таким образом, для получения единственного, стереоизомерно чистого продукта приходится решать задачу стереоселективности .
Следует отметить, что дифференцировать энантиомеры, принадлежащие одному и тому же диастереомеру весьма затруднительно, поскольку переходные состояния , ведущие к этим энантиомерам, также энантиомерны и поэтому в ахиральной среде совершенно одинаковы по энергии. Для того, чтобы дифференциация стала осуществима, обычно в структуру реагентов вводят какой-либо хиральный фрагмент, чтобы сделать переходные состояния диастереомерными и неравнозначными .
Если альдегид уже содержит стереоцентр с известной конфигурацией , то альдольная конденсация с образованием второго стереоцентра будет протекать с той или иной диастереоселективностью, то есть вновь создаваемый стереоцентр будет преимущественно представлен одной преобладающей конфигурацией. Предсказать и объяснить стереохимический результат такого превращения можно при помощи стандартных моделей, которые используются в стереохимии для реакций нуклеофильного присоединения к карбонильной группе (например, модели Фелкина — Ана , модели с хелатированием и т. д.) .

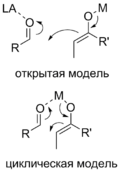
Для предсказания стереохимического результата реакции ахирального енолята с ахиральным альдегидом, в которой происходит образование двух стереоцентров, применяют две модели переходного состояния : открытую и циклическую. Открытая стереохимическая модель предусматривает относительно большую подвижность молекул, в первую очередь, вращение вокруг нескольких одинарных связей. Поэтому реакции, протекающие согласно такой модели, имеют низкую диастереоселективность. Напротив, шестичленное переходное состояние в циклической модели (модели Циммермана — Тракслера) имеет жёсткую структуру, более ярко проявляющиеся пространственные взаимодействия и, соответственно, лучше передаёт стереохимическую информацию, что в итоге выражается в высокой селективности .
Переходное состояние в модели Циммермана — Тракслера представляет собой циклический шестичленный комплекс карбонильного соединения и енолята в наиболее устойчивой конформации «кресло». Из возможных вариантов расположения молекул при этом реализуется тот, при котором пространственные затруднения минимальны. Так, для E -енолята реализуется два переходных состояния, однако одно из них менее устойчиво, поскольку в нём присутствуют невыгодные между заместителями R и X. Соответственно, продукт с син -расположением заместителей, к которому ведёт данное переходное состояние, образуется в меньшем количестве, а основным является анти -продукт . Аналогично, для Z -енолята более выгодным является образование син -продукта. Таким образом, в данном типе альдольных реакций стереохимический результат определяется конфигурацией двойной связи енолята .
Модель Циммермана — Тракслера реализуется при наличии у атома металла в еноляте способности координироваться с карбонильной группой альдегида. При этом чем короче связь металл — кислород, тем более компактным является переходное состояние и тем более высока стереоселективность. С этой точки зрения, наиболее выгодно использовать еноляты титана и бора .


Данные соображения применимы в случае кинетического контроля, когда реакция протекает быстро и необратимо. Если же реализуется термодинамический контроль с установлением равновесия на стадиях процесса, то из енолятов любой конфигурации образуется анти -изомер как более устойчивый, поскольку в переходном состоянии большее число заместителей находится в экваториальном положении .
На стереохимию процесса оказывает влияние также размер заместителя R в альдегиде и заместителя X в еноляте. Если эти группы имеют большой объём ( трет -бутил, неопентил и др.), то реакция протекает с высокой стереоизбирательностью. В то же время при X небольшого объёма (этил, изопропил, трет -бутокси, диизопропиламино) стереоселективность понижается или исчезает .
Как показано выше, диастереоселективность реакций альдольной конденсации достигается за счёт использования енолятов той или иной конфигурации. Преимущественное же образование одного из энантиомеров (энантиоселективность) достигается при использовании енолятов, содержащих стереоцентр определённой конфигурации. При этом для предсказания абсолютной конфигурации получаемого продукта также используются шестичленные модели переходного состояния. Предполагается, что более выгодным является такое переходное состояние, в котором взаимодействие аксиального атома водорода с самым объёмным заместителем хирального атома (R L ) минимально (в конечном итоге будет образовываться продукт с син -расположением метильной группы относительно наименьшего заместителя R S ) .

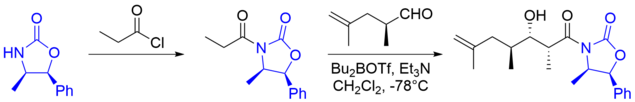
Далеко не всегда в структуре енолята имеется направляющий хиральный центр, поэтому были разработаны методики введения в еноляты . В частности, одним из самых успешных подходов является методология Эванса . Она заключается в использовании карбонильных соединений, содержащих хиральный фрагмент. Такие вещества образуют преимущественно Z -еноляты (лучшие результаты дают еноляты бора , титана и олова ) и затем — при реакции с альдегидом — син -альдоли, согласно модели Циммермана — Тракслера. Диастереоселективность для некоторых случаев превышает 99 %. Оксазолидины затем можно гидролизовать до карбоновых кислот или превратить в .
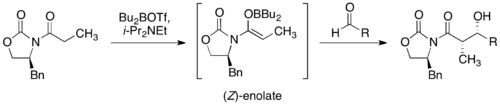
Использование хиральных вспомогательных реагентов имеет существенный недостаток: требуются дополнительные стадии синтеза, на которых вводится, а затем удаляется хиральный фрагмент, что снижает общий выход продукта. Кроме того, сам хиральный индуктор необходим в стехиометрическом количестве. В связи с этим разрабатываются методики, включающие использование металлоорганического реагента с хиральным лигандом на стадии получения енолята либо хиральных катализаторов .
Подход с использованием хиральных катализаторов является наиболее привлекательным, поскольку в нём стереоконтроль осуществляется при помощи небольшого (каталитического) количества хирального материала, что обеспечивает гораздо большую экономию по сравнению с методами, где хиральные соединения используются в стехиометрическом количестве. Наибольшие возможности для поиска хиральных катализаторов предоставляет , для проведения которой требуется внешний реагент — кислота Льюиса. Если использовать хиральную кислоту Льюиса, можно добиться того, что хиральность в продукт будет привноситься из этой кислоты. Такие кислоты были найдены. Первый общий метод энантиоселективной реакции Мукаямы был основан на использовании хирального комплекса титана в качестве кислоты Льюиса, причём его расход был равен 2—5 мольным процентам, а энантиоселективность достигала 94 % (то есть соотношение энантиомеров составляло 97 : 3) .

В последнее время особое внимание уделяется реакциям, в которых используется минимальное количество вспомогательных реагентов (концепция). Альдольная конденсация, с этой точки зрения, должна проводиться по классическим методикам, под действием кислоты или щёлочи, без предварительного генерирования енолятов, но в энантиоселективном варианте. Такие реакции называют прямыми альдольными реакциями ( англ. direct aldol reactions), в отличие от направляемых альдольных реакций ( англ. direct ed aldol reactions) с генерированием енолятов .
Оптимальным катализатором, который был применён во многих стереоселективных альдольных конденсациях, стал L- пролин . Его действие связано с одновременным наличием в его молекуле двух функциональных групп: амина и карбоновой кислоты . Амин образует енамин с метиленовой компонентой, активируя её, а карбоксильная группа активирует карбонильную компоненту. Стереоконтроль в реакции осуществляется за счёт наличия в пролине хирального центра. Очевидно, что карбонильные соединения должны заметно отличаться по реакционной способности, чтобы перекрёстная конденсация прошла с образованием одного продукта .

С точки зрения стереохимии реакции, образующиеся α,β-ненасыщенные карбонильные соединения могут иметь различную конфигурацию двойной связи . Имеющиеся примеры показывают, что более выгодным и устойчивым является транс -изомер (с транс -расположением большего β-заместителя и карбонильной группы). Цис -изомеры под действием кислот или оснований изомеризуются в транс -продукты. Обратное превращение достигается при облучении ультрафиолетовым излучением .
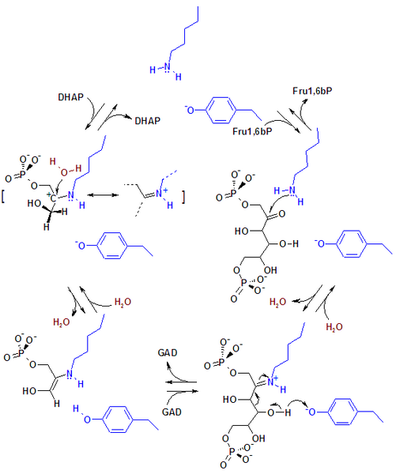
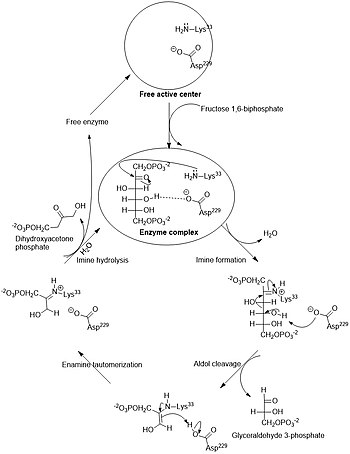
Альдольные реакции протекают во многих метаболических путях, но наиболее распространены они в метаболизме углеводов, где катализируются альдолазами — ферментами , принадлежащими к классу лиаз . С точки зрения механизма этот катализ осуществляется двумя путями. Альдолазы I типа обнаруживаются преимущественно в высших растениях и животных. Они действуют по енаминному механизму, согласно которому лизиновый остаток фермента генерирует енамин B в активном сайте. Енамин затем атакует альдегид ( С ), образуя аддукт D . Затем после гидролиза образуется альдоль .
Альдолазы II типа находятся в бактериях и грибах и используют ион Zn 2+ в качестве кофактора . Этот ион располагается между двумя субъединицами гомотетрамерного белка и активирует метиленовую компоненту за счёт координации с двумя атомами кислорода и образования нуклеофильного ендиолята F . Альдолаза также активирует карбонильную компоненту через образование водородной связи . После атаки енолята на карбонильную компоненту происходит разрушение комплекса и образование альдоля .

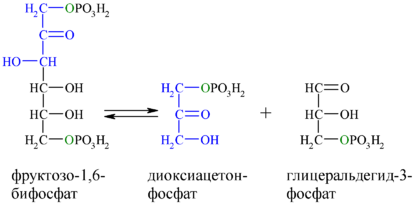
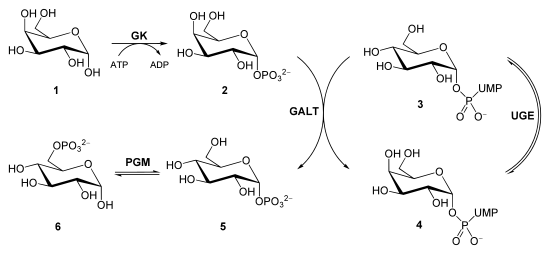
И альдольная, и ретроальдольная реакции входят в метаболические пути углеводов: обе реакции могут участвовать в создании или разрушении сахаров в зависимости от потребностей клетки. Так, последовательность стадий биосинтеза глюкозы ( глюконеогенез ) включает в себя реакцию дигидроксиацетонфосфата , выполняющего роль нуклеофила , и глицеральдегид-3-фосфата ( электрофила ) с образованием фруктозо-1,6-дифосфата . Данная реакция также катализируется альдолазой и протекает региоселективно , несмотря на обилие функциональных групп в субстратах. В ходе гликолиза происходит обратная реакция, являющаяся по сути ретроальдольной .

В цикле Кребса присутствует подобная реакция, протекающая между ацетил-коферментом А и оксалоацетатом в присутствии фермента цитратсинтазы . В данном случае ацетил-КоА является метиленовой компонентой, а оксалоацетат — карбонильной компонентой. Фермент также катализирует разложение тиоэфира, за счёт чего продуктами реакции являются лимонная кислота и кофермент А .

Ряд альдолаз, а также антитела , имитирующие их действие, но имеющие бо́льшую субстратоспецифичность, используются для проведения альдольных конденсаций в мягких условиях, близких к физиологическим . Механизм действия альдолаз также стимулирует поиск новых низкомолекулярных катализаторов, действующих по похожему принципу, однако, превалирующим подходом остаётся получение енолята в отдельную стадию .
Альдольная реакция является одной из важнейших реакций в синтезе природных соединений. Это связано с её возможностями по направленному созданию хиральных центров. Кроме того, целый класс природных соединений — поликетидов — включает 1,3-кислородсодержащие фрагменты, поэтому альдольная реакция играет ключевую роль в их синтезе .
Альдольная конденсация применяется в промышленном синтезе бутанола-1 , 2-этилгексанола и пентаэритрита .