Хронический тонзиллит
- 1 year ago
- 0
- 0

Хронический лимфолейкоз , или хронический лимфоцитарный лейкоз (ХЛЛ) , — злокачественное клональное лимфопролиферативное заболевание, характеризующееся накоплением атипичных зрелых CD5 / CD19 / CD23 -положительных В-лимфоцитов преимущественно в крови, костном мозге , лимфатических узлах , печени и селезёнке .
Хронический лимфолейкоз — одно из наиболее распространённых онкогематологических заболеваний . Также это наиболее частый вариант лейкоза среди представителей европеоидной расы . По непонятным причинам редко встречается среди населения стран Восточной Азии . Дебют заболевания, как правило, происходит в пожилом возрасте — медианный возраст на момент постановки диагноза составляет 70—72 года. Мужчины болеют в 1,5—2 раза чаще, чем женщины. Ежегодная заболеваемость составляет 6,8 случая на 100 тыс. мужчин и 3,5 случая на 100 тыс. женщин .
Предрасположенность к заболеванию передаётся по наследству — риск развития хронического лимфоцитарного лейкоза у непосредственных родственников в 8,5 раз превышает популяционный, однако даже при этом остается ниже 1 % . Описаны семейные случаи с относительно высокой пенетрантностью. Большей части случаев ХЛЛ, если не всем, предшествует предлейкозное состояние (моноклональный В-клеточный лимфоцитоз), которое наблюдается у 5—10 % людей в возрасте старше 40 лет и прогрессирует в ХЛЛ с частотой около 1 % в год .
Анализ генома людей с наследственным ХЛЛ позволил идентифицировать однонуклеотидные полиморфизмы , ассоциированные с этим состоянием. Полиморфизмы были обнаружены примерно в 30 локусах , например, в генах IRF4 , и BCL2 .
Контакт с агентом «оранж» и инсектицидами могут повышать риск развития ХЛЛ.
Этиологическая связь ХЛЛ с ионизирующим излучением , вирусными инфекциями, питанием и образом жизни не доказана .
Первоначально хронический лимфоцитарный лейкоз рассматривали как онкологическое заболевание, характеризующееся накоплением долгоживущих, но очень редко делящихся иммунологически некомпетентных B-лимфоцитов . Однако исследования с использованием тяжёлой воды показали, что злокачественные клетки пролиферируют , и достаточно быстро — количество новых клеток, образующихся за день, составляет от 0,1 до более чем 1 % от общего числа клеток клона . Причём при высокой скорости пролиферации более вероятно агрессивное течение болезни.
Клеточное микроокружение (ниша) играет большую роль в патогенезе хронического лимфолейкоза. Пролиферация злокачественных клеток происходит в микроанатомических структурах, которые называются пролиферативными центрами, или псевдофолликулами. Псевдофолликулы представляют собой скопления лейкозных клеток, находящихся в контакте со вспомогательными клетками (например, стромальными клетками), которые стимулируют их пролиферацию и выживание . Пролиферативные центры в основном находятся в лимфатических узлах и в меньшей степени в костном мозге .
Злокачественные клетки имеют CD19/CD5/CD23-положительный иммунофенотип и низкий уровень мембранных иммуноглобулинов . Нормальные популяции В-клеток с таким набором поверхностных маркеров неизвестны, что мешает установить, какой тип клеток может давать начало злокачественному клону при ХЛЛ. Анализ транскриптома показал, что опухолевые клетки по набору синтезируемых мРНК похожи на зрелые В-клетки, которые прошли активацию антигеном . В норме таким профилем экспрессии генов обладают В-клетки памяти и В-клетки краевой зоны лимфатических фолликулов , поэтому предполагают, что именно они могут быть предшественниками лейкозных клеток .
В отличие от других B-клеточных лейкозов, для ХЛЛ не удалось выявить типичных хромосомных транслокаций , затрагивающих онкогены . Кроме того, крупные хромосомные перестройки редко наблюдаются на ранних стадиях заболевания, так что маловероятно, что они являются первичной причиной ХЛЛ. Однако по мере прогрессирования заболевания такие перестройки происходят: чаще всего это делеции участков хромосом 11 , 13 и 17 .
Характерен абсолютный лимфоцитоз в периферической крови (по данным гемограммы ) и костном мозге (по данным ). На ранних стадиях лимфоцитоз является единственным проявлением заболевания. Пациенты могут предъявлять жалобы на так называемые «конституциональные симптомы» — астению , повышенную потливость, спонтанное снижение массы тела.
Характерна генерализованная лимфаденопатия . Увеличение внутригрудных и внутрибрюшных лимфатических узлов выявляется при ультразвуковом или рентгенологическом обследовании, периферические лимфоузлы доступны пальпации. Лимфатические узлы могут достигать значительных размеров, образовывать мягкие или плотноватые конгломераты. Сдавление внутренних органов не характерно.
На более поздних стадиях заболевания присоединяется гепатомегалия и спленомегалия . Увеличение селезёнки может проявляться ощущением тяжести или дискомфорта в левом подреберье, феноменом раннего насыщения.
За счёт накопления опухолевых клеток в костном мозге и вытеснения нормального гемопоэза на поздних стадиях могут развиваться анемия , тромбоцитопения , редко . Поэтому пациенты могут жаловаться на общую слабость, головокружения, петехии, экхимозы, спонтанную кровоточивость.
Анемия и тромбоцитопения также могут иметь аутоиммунный генез .
Для заболевания характерна выраженная иммуносупрессия , затрагивающая преимущественно гуморальный иммунитет ( ). Из-за этого имеется предрасположенность к инфекциям, например, рецидивирующим простудным заболеваниям и пневмонии.
Необычным клиническим проявлением заболевания может быть гиперреактивность на укусы насекомых.
Для дифференциальной диагностики хронического лимфоцитарного лейкоза с другими лимфопролиферативными заболеваниями необходимо проанализировать количество В-клеток в периферической крови, мазок крови и провести иммунофенотипирование циркулирующих в крови лимфоцитов. Дополнительно для определения прогноза (но не схемы лечения) иногда проводят цитогенетическое исследование , определяют мутационный статус локуса IgV H , количество или CD38 в клетках ХЛЛ, количество CD23, тимидинкиназы и β 2 -микроглобулина в сыворотке крови, а также анализируют биоптат или аспират костного мозга .

Необходимым критерием диагноза хронического лимфоцитарного лейкоза является повышение абсолютного числа В-лимфоцитов в крови до или более 5×10 9 /л. Кроме того, эти лимфоциты должны иметь характерный иммунофенотип: на их поверхности должны обнаруживаться CD19 , CD5 , CD23 , небольшие количества CD20 и , а также лёгкие цепи иммуноглобулинов .
В мазке крови обнаруживаются опухолевые клетки, которые имеют морфологию зрелых (малых) лимфоцитов: «штампованное» ядро с конденсированным хроматином без ядрышка , узкий ободок цитоплазмы. Характерно наличие так называемых теней Гумпрехта, которые представляют собой лейкозные клетки, разрушившиеся в процессе приготовления мазка. Помимо малых лимфоцитов в мазке могут присутствовать более крупные или атипичные клетки, иногда отмечается существенная (более 10 %) примесь омоложённых клеток (пролимфоцитов и параиммунобластов), требующая проведения дифференциального диагноза с пролимфоцитарным лейкозом .
Иммунофенотипирование лимфоцитов методом проточной цитометрии обязательно для подтверждения диагноза. Высокочувствительная проточная цитометрия позволяет обнаруживать одну злокачественную клетку на 10 000 нормальных лейкоцитов . В качестве диагностического материала обычно используется периферическая кровь. Для клеток ХЛЛ характерен аберрантный иммунофенотип: одновременная экспрессия (коэкспрессия) Т-клеточного маркера CD5 и В-клеточных маркеров CD19 и CD23 . Количество В-клеточных маркеров CD20 , и мембраносвязанных иммуноглобулинов IgM и IgD понижено по сравнению с нормальными В-клетками . В дополнение к этому выявляется . Диагноз ХЛЛ также может быть установлен на основании данных иммуногистохимического исследования биоптата лимфатического узла или селезёнки.
Подозрение на хронический лимфоцитарный лейкоз также возникает в случае обнаружения у в остальном здоровых людей увеличения абсолютного числа клональных B-лимфоцитов соответствующего иммунофенотипа, даже если общее их количество в периферической крови меньше 5000/микролитр. Если этому признаку не сопутствует лимфаденопатия или органомегалия, цитопении или другие признаки заболевания, такое состояние диагностируется как моноклональный B-лимфоцитоз . Согласно исследованию, проведённому на 1520 участниках в возрасте от 62 до 80 лет с нормальными показателями крови, моноклональный B-лимфоцитоз с иммунофенотипом ХЛЛ обнаруживается у 5 % людей в этой возрастной группе. Такой лимфоцитоз может прогрессировать в ХЛЛ со скоростью около 1 % в год .
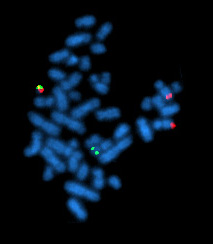
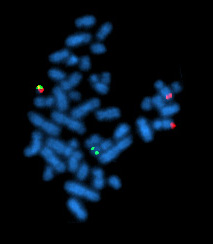
Цитогенетическое исследование проводится методом стандартного кариотипирования или FISH . Задача исследования — выявление хромосомных мутаций , часть из которых имеет прогностическую значимость. Из-за возможности клональной эволюции исследование должно повторяться перед каждой линией терапии и в случае возникновения рефрактерности.
Стандартное кариотипирование возможно только для клеток в метафазе клеточного цикла . Так как злокачественные клетки при ХЛЛ обладают низкой митотической активностью, для получения необходимого для анализа количества метафаз требуется применение митогенов . Но даже в таком случае хромосомные мутации удаётся обнаружить только в 40—50 % случаев .
Интерфазная FISH при хроническом лимфоцитарном лейкозе не требует применения митогенов и отличается большей чувствительностью. При анализе используют локус-специфичные зонды, позволяющие выявлять наиболее распространённые хромосомные перестройки (как правило делеции ). Этот метод позволяет детектировать хромосомные мутации в более чем 80 % случаев хронического лимфоцитарного лейкоза .
У каждого отдельного пациента может быть обнаружена одна, две и более стандартных мутации. Исследование, проведённое на 325 пациентах с хроническим лимфоцитарным лейкозом, позволило установить иерархию кариопитов по их прогностической способности: del17p, del11q, трисомия 12, нормальный кариотип и del13q. Если у пациента обнаружено больше одной мутации, то прогноз делают по той из них, которая находится выше в этом списке .

Хромосомные перестройки ассоциированы с определёнными клиническими характеристиками заболевания :
Рутинный физикальный осмотр позволяет получить достаточное представление о клинической динамике, поскольку заболевание носит системный характер. Выполнение УЗИ и компьютерной томографии для оценки объёма внутренних лимфоузлов не является обязательным вне клинических исследований.
Тест на гемолитическую анемию из-за высокой частоты аутоиммунных осложнений при ХЛЛ необходим даже при отсутствии её явных клинических проявлений. Рекомендуется проводить прямую , подсчёт числа ретикулоцитов и определение уровня фракций билирубина .
Как правило проведение биопсии костного мозга не требуется для поставки диагноза. Анализ биоптата может помочь сделать прогноз относительно скорости развития болезни, но последние наблюдения показывают, что использование других прогностических маркеров может давать лучшие результаты. Однако анализ аспирата или пунктата костного мозга может понадобиться для выяснения причин цитопении (специфическое поражение костного мозга или аутоиммунное осложнение) путём исследования .
Некоторые дополнительные тесты используются для предсказания скорости прогрессирования заболевания, но не влияют на выбор схемы лечения. К таким тестам относится определение наличия соматических мутаций в вариабельной области генов тяжёлых цепей иммуноглобулинов (IgV H ) и определение количества CD38 и ZAP-70 в клетках, поражённых хроническим лимфоцитарным лейкозом. IgV H без мутаций указывают для более агрессивную болезнь и менее благоприятный прогноз . Экспрессия CD38 и ZAP-70 коррелирует с отсутствием мутаций в IgV H и плохим прогнозом. Однако пока не до конца ясно, являются ли эти молекулы независимыми прогностическими факторами . Также на агрессивность болезни указывают повышенное количество тимидинкиназы , CD23 и β2-микроглобулина в сыворотке крови.
Используются системы стадирования, предложенные K. Rai и J. Binet . Оригинальная система Rai была модифицирована с целью снизить количество определяемых групп риска с 5 до 3 . Обе системы опираются на данные физического осмотра и стандартные лабораторные анализы и легки в применении. В них отражено естественное течение заболевания — постепенное накопление опухолевой массы. Стадирование позволяет делать прогнозы о выживаемости: прогноз пациентов на поздних стадиях может быть хуже, чем на более ранних. Однако эти системы не дают возможности прогнозировать индивидуальный риск прогрессирования заболевания и выживания на ранних стадиях (стадии 0-II по Rai, A по Binet) . По этой причине стали широко использовать другие прогностические маркеры, такие как цитогенетические характеристики злокачественных клонов, мутационный статус локуса IgV H и количество или CD38 .
|
|
|||||||||||||||||||||||
Хронический лимфолейкоз является практически неизлечимым медленнопрогрессирующим (индолентным) заболеванием.
Лечение не начинается сразу после подтверждения диагноза. Заболевание может сохранять стабильность годами, иногда в течение всей жизни больного. Часто наблюдается волнообразное течение с периодами увеличения и уменьшения опухолевого объема. Решение о необходимости начала терапии принимается обычно после периода более или менее длительного наблюдения.
Показания для начала лечения сформулированы в современных рекомендациях. Они отражают картину активной прогрессии заболевания, приводящей к ухудшению медицинского состояния больного и/или качества его жизни.
Из-за системного характера заболевания радиотерапия при хроническом лимфоцитарном лейкозе не применяется. Стандартом терапии являются химиотерапевтические режимы с включением нуклеотидных аналогов, алкилирующих препаратов и моноклональных антител.
Один из наиболее эффективных режимов — «FCR» ( англ. fludarabine, cyclophosphamide, rituximab ). Он позволяет получить полную ремиссию примерно у 85 % больных низкой группы риска. Однако этот режим имеет побочные эффекты, которые не позволяют использовать его для пациентов пожилого возраста. Кроме того, режим малоэффективен для больных группы высокого риска (например, имеющих делецию 17p) .
| Режим FCR | |
|---|---|
| Флударабин | 25 мг/м 2 в/в или 40 мг/м 2 р.о. 1—3 дни |
| Циклофосфамид | 250 мг/м 2 в/в 1—3 дни |
| Ритуксимаб | 375 мг/м 2 (1 курс) или 500 мг/м 2 (2—6 курсы) 1 или 0 день |
Активно исследуется возможность применения в терапии алкилирующего препарата .
Резистентость к цитостатикам, как правило, обусловлена нарушением механизмов инициации апоптоза в ответ на повреждения ДНК в клетках опухоли. Наиболее типичны мутации гена TP53 , приводящие к его инактивации. Клетки с инактивированным p53 не погибают при накоплении повреждений генома. Более того, мутации, индуцированные цитостатиками, могут давать таким клеткам дополнительное преимущество за счет активации онкогенов или инактивации антионкогенов . Таким образом, мутагенез, индуцированный цитостатиками, может являться двигателем клональной эволюции.
У пациентов с резистентным течением в настоящее время используются высокие дозы глюкокортикостероидов, алемтузумаб (моноклональное антитело к CD52 ), режимы, его содержащие, а также аллогенная трансплантация костного мозга .
Проведение интенсивной химиотерапии и трансплантации костного мозга у пожилых может быть затруднено плохим соматическим статусом и наличием серьезных сопутствующих заболеваний. В этой группе больных часто используется хлорамбуцил или комбинации на его основе.
Новые препараты ( леналидомид , BGB-3111, , дувелисиб , ) и комбинированные режимы на их основе в настоящее время проходят заключительные этапы клинических испытаний.
Существует также значительное количество новых экспериментальных подходов к терапии хронического лимфоцитарного лейкоза, эффективность и безопасность которых окончательно не установлена.
В последние годы показана высокая эффективность ингибиторов тирозинкиназ Btk ( ибрутиниб , и др.) и PI3Kdelta ( иделалисиб и др.), а также высокоселективного ингибитора Bcl-2 ( венетоклакс ). В 2014 году FDA (Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США) выдало разрешение на применение ибрутиниба у больных ХЛЛ, предварительно прошедших как минимум один курс лечения . Данные таргетные препараты обладают высокой активностью даже у пациентов с неблагоприятным прогнозом (del17p) и относительно малотоксичны. В то же время, недостатком является их крайне высокая стоимость.
Моноклональные антитела : офатумумаб , обинутузумаб , моксетумомаб пасудотокс .
По клиническим проявлениям хронический лимфоцитарный лейкоз является довольно гетерогенным заболеванием: болезнь может протекать длительно без прогрессии или, наоборот, очень агрессивно . Примерно в 30 % случаев болезнь прогрессирует медленно, так что смерть наступает по причине, не связанной с болезнью. В 15 % случаев смерть от болезни и/или побочных эффектов лечения наступает в течение 2—3 лет с момента постановки диагноза. В остальных случаях болезнь медленно прогрессирует в течение 5—10 лет, после чего наступает терминальная стадия заболевания, за которой следует смерть . В случае пациентов из группы низкого риска медиана выживаемости от момента постановки диагноза достигает 8—10 лет. Известен ряд факторов, которые позволяют прогнозировать результаты лечения и продолжительность жизни, в том числе:
Опухолевая трансформация, при которой клетки клона приобретают новые характеристики, делающие их похожими на диффузную крупноклеточную лимфому , носит название синдром Рихтера. Прогноз при наличии трансформации крайне неблагоприятный.
| Кроветворение | |
|---|---|
| Компоненты | |
| Биохимия | |
| Заболевания | |
|
См. также:
Гематология
,
Онкогематология
|
|
| Топография | |
|---|---|
| Морфология |
|
| Лечение | |
| Родственные структуры | |
| Прочее | |