Interested Article - Болезнь Гентингтона

- 2020-12-03
- 1
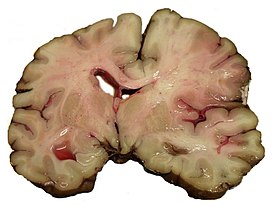
Болезнь Хантингтона ( синдром Гентингтона , хорея Гентингтона , хорея Хантингтона ) — аутосомно-доминантное генетическое заболевание нервной системы, характеризующееся постепенным началом обычно в возрасте 30—50 лет и сочетанием прогрессирующего хореического гиперкинеза и психических расстройств. Заболевание вызывается умножением кодона CAG в гене HTT. Этот ген кодирует 350-kDa белок гентингтин с неизвестной функцией. В гене дикого типа (не мутантного) у разных людей присутствует разное количество CAG-повторов, однако, когда число повторов превышает 36, развивается болезнь. Нейроморфологическая картина характеризуется атрофией стриатумa , а на поздней стадии также атрофией коры головного мозга.
Эпидемиология
В настоящее время от хореи Гентингтона в США страдает около 7000 человек. Частота встречаемости заболевания среди населения с европейскими корнями составляет примерно 3–7:100000 и 1:1000000 среди остальных рас [ нет в источнике ] . Название болезни дано в честь трёх поколений врачей, изучавших её в штате Коннектикут . В частности, считается, что заболевание названо в честь американского врача Джорджа Хантингтона (Гентингтона), первым давшего его классическое описание .
Генетика
Ген HTT , присутствующий у всех людей, кодирует белок гентингтин (Htt). Ген HTT расположен на коротком плече 4-й хромосомы (4p16.3) . Этот ген содержит в себе участок с повторяющейся последовательностью трёх азотистых оснований — цитозин - аденин - гуанин (то есть, ЦАГЦАГЦАГ…). Триплет ЦАГ кодирует аминокислоту глутамин , поэтому синтезируемый белок гентингтин содержит последовательность глутаминовых аминокислот, называемую полиглутаминовый тракт .
Количество ЦАГ триплетов различно у отдельных лиц и может изменяться с последующими поколениями. Если их становится больше 36, то синтезируется удлинённый полиглутаминовый тракт и происходит образование мутантного белка гентингтина (mHtt) , который оказывает токсичное действие на клетки и вызывает болезнь Гентингтона. Как правило, от числа ЦАГ повторов зависит степень повреждений, наличие около 60 % повторов сверх нормы вызывает появление симптомов в различном возрасте . 36—40 повторов приводят к редуцированной пенетрантности формы этого заболевания, которая намного позже проявляется и медленнее прогрессирует. В некоторых случаях начало болезни может быть настолько поздним, что симптомы никогда не обнаруживаются . При очень большом количестве повторов болезнь Гентингтона имеет полную пенетрантность и может проявиться до 20 лет, тогда болезнь классифицируется как ювенильная, акинетически-ригидная или Вестфаль варианты. Составляет приблизительно 7 % случаев болезни Гентингтона .
Мутантный ген был предположительно завезён в США в 1630 году двумя братьями, эмигрировавшими из Эссекса в Бостон .
Болезнь передаётся по наследству. Мутантный аллель доминантный, поэтому в семье, где один из родителей несёт такую мутацию, каждый из потомков может получить её с вероятностью 50 %. Наследование не зависит от пола носителя или его детей.
Патогенез
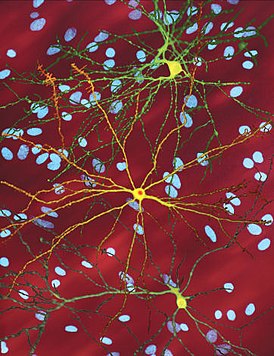
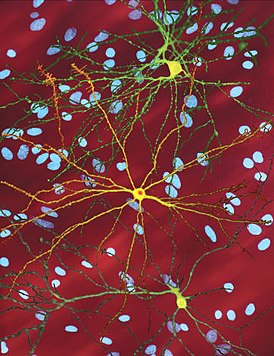
Htt-белок взаимодействует с сотней других белков и, вероятно, выполняет множество биологических функций . Механизм действия mHtt до конца не ясен, но известно, что он токсичен для некоторых типов клеток, особенно в головном мозге. В основном происходит поражение полосатого тела ( стриатума ), но при прогрессировании заболевания и другие области головного мозга значительно повреждаются . Планирование и коррекция движений — основная функция полосатого тела, и нарушения в этой области провоцируют симптомы .
Функция Htt
Htt образуется во всех клетках млекопитающих. Наибольшая его концентрация — в головном мозге и яичках , а также в умеренных количествах в печени , сердце и лёгких . Функция Htt у человека не ясна. Он взаимодействует с белками, участвующими в транскрипции , передаче сигнала в клетке и внутриклеточном транспорте . Некоторые функции Htt обнаружены в экспериментальных моделях животных: играет важную роль в развитии эмбриона и связан с гибелью эмбриона при отсутствии белка . Он также выступает в качестве анти- апоптозного агента, предотвращая запрограммированную гибель клеток, и контролирует образование нейротрофического фактора мозга (белок, защищающий нейроны и регулирующий их образование во время нейрогенеза ). Если экспрессия Htt возрастает, выживаемость нервных клеток увеличивается и эффект mHtt уменьшается, наоборот, понижение экспрессии Htt даёт картину более типичную присутствию mHtt . У людей разрушение нормального гена не приводит к болезни. В настоящее время считается, что болезнь вызывает не недостаточное образование Htt, а усиление токсического эффекта mHtt .
Клеточные изменения под действием mHtt
Под действием образовавшегося mHtt происходит множество изменений в клетке, что вызывает болезнь Гентингтона. Удлинение полиглутаминовой последовательности изменяет конформацию белка гентингтина и прочно соединяет его с другими белками . Это приводит к агрегации гентингтина, при этом образуются так называемые внутриклеточные тельца включения . Эти включения механически препятствуют движению везикул , содержащих нейромедиаторы , через цитоскелет , что нарушает передачу сигналов в нейронах . Тельца включения обнаруживаются как в ядрах клеток , так и в цитоплазме . Некоторые эксперименты показали, что они могут быть токсичны для клеток, а другие — что тельца, наоборот, защищают нейрон от смерти, аккумулируя мутантный гентингтин, и именно неагрегированный белок токсичен .
Существует несколько путей, при которых mHtt вызывает гибель клеток. К ним относят: влияние на белки- шапероны ; взаимодействие с каспазами , которые участвуют в апоптозе; токсическое действие глутамина на нервные клетки ; нарушение выработки энергии в клетках и влияние на экспрессию генов. Токсическое действие mHtt значительно усиливается при взаимодействии с белком RASD2 (Rhes), который образуется преимущественно в стриатуме . RASD2 вызывает сумоляцию ( SUMOylation ) mHtt к образованию белковых сгустков и дезагрегации — исследования в культуре клеток показали, что сгустки менее токсичны, чем дезагрегированная форма .
Макроскопические изменения под действием mHtt

Болезнь Гентингтона поражает специфические области мозга. Наиболее заметные ранние изменения затрагивают область базальных ганглиев , называемую полосатым телом , которое состоит из хвостатого ядра и скорлупы . Другие повреждаемые области включают чёрную субстанцию , 3, 5 и 6 слои коры головного мозга , гиппокамп , клетки Пуркинье в мозжечке , боковые туберальные ядра гипоталамуса и часть таламуса . Эти области получают повреждения в соответствии с их структурой и типами содержащихся в них нейронов, уменьшаясь в размерах в связи с гибелью клеток . Звёздчатые нейроны полосатого тела наиболее уязвимы, особенно проецирующиеся в направлении поверхности бледного шара , вставочные и звёздчатые нейроны, проецирующиеся к центру бледного шара, получают меньше повреждений . Болезнь Гентингтона также вызывает аномальное увеличение астроцитов .
Базальные ганглии — часть головного мозга, наиболее заметно повреждающиеся при болезни Гентингтона — играют ключевую роль в контроле движений и поведения. Их функция полностью не ясна, но современные теории предполагают, что они являются частью когнитивной исполнительной системы . Базальные ганглии в норме ингибируют большое число контуров ( circuit ), генерирующих специфические движения. Для инициации специфических движений кора посылает сигналы базальным ганглиям для снятия ингибирования. Повреждение базальных ганглиев может привести к снятию ингибирования или его постоянным неконтролируемым изменениям, что служит причиной затруднения начала движения или к их непроизвольной инициации, или движение может быть прервано до или после достижения желаемого результата. Накапливающиеся повреждения в этой области приводят к беспорядочным движениям, характерным для болезни Гентингтона .
Симптоматика
Симптомы болезни Гентингтона могут проявиться в любом возрасте, но чаще это происходит в 35—44 года . На ранних стадиях происходят небольшие изменения личности, когнитивных способностей и физических навыков . Обычно первыми обнаруживают физические симптомы, так как когнитивные и психические расстройства не столь выражены в ранних стадиях . Почти у всех пациентов болезнь Гентингтона в итоге проявляется схожими физическими симптомами, но начало заболевания, прогрессирование и степень когнитивных и психических нарушений различаются у отдельных лиц .
Для начала заболевания наиболее характерна хорея — беспорядочные, неконтролируемые движения. Хорея вначале может проявляться в беспокойстве, небольших непроизвольных или незавершённых движениях, нарушении координации и замедлении скачкообразных движений глаз .
В самом начале обычно возникают проблемы из-за физических симптомов, которые выражаются в резких, внезапных и не поддающихся контролю движениях. В других случаях, наоборот, больной двигается слишком замедленно. Возникают нарушения координации движений, речь становится невнятной. Постепенно все функции, требующие мышечного контроля, нарушаются: человек начинает гримасничать, испытывает проблемы с жеванием и глотанием. Из-за быстрого движения глаз происходят нарушения сна. Обычно больной проходит через все стадии физического расстройства, однако влияние болезни на когнитивные функции у всех очень индивидуально. Чаще всего происходит расстройство абстрактного мышления, человек перестаёт быть способным планировать свои действия, следовать правилам, оценивать адекватность своих действий. Постепенно появляются проблемы с памятью, могут возникнуть депрессия и паника, эмоциональный дефицит, эгоцентризм , агрессия , навязчивые идеи, проблемы с узнаванием других людей, гиперсексуальность и усиление вредных привычек, таких как алкоголизм или игромания . Бывает, что человек, болеющий хореей Гентингтона, не понимает, сыт он или голоден. Тогда больных приходится кормить 3—4 раза в день и не перекармливать.
Диагностика
Клинические методы
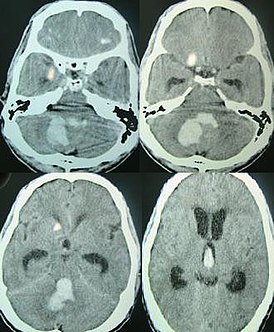
Физикальное обследование , иногда в сочетании с психологическим обследованием, позволяет определить область распространения болезни . Медицинская визуализация ( компьютерная томография (КТ), магнитно-резонансная томография (МРТ)) показывает только видимую атрофию мозга на прогрессирующей стадии заболевания. Методы функциональной нейровизуализации ( фМРТ и позитронно-эмиссионная томография (ПЭТ)) могут показать изменения в активности мозга до появления клинических симптомов .
Генетические методы
Для проведения генетической диагностики болезни Гентингтона необходим забор крови с последующим определением количества повторов ЦАГ в каждом НТТ -аллеле . Положительный результат не подтверждает диагноз, поскольку может быть получен за несколько лет до появления первых симптомов. Однако отрицательный результат однозначно свидетельствует об отсутствии вероятности развития болезни Гентингтона .
Эмбрионы
Эмбрионы , полученные в результате экстракорпорального оплодотворения , могут быть подвержены генетической диагностике болезни Гентингтона с применением преимплантационной генетической диагностики . При этом методе забирается одна клетка из 4-8-клеточного эмбриона и затем проверяется на генетическую патологию. Полученная информация может впоследствии быть использована при выборе здорового эмбриона для имплантации. Кроме того, возможна пренатальная диагностика для эмбриона или плода в утробе матери .
Дифференциальная диагностика
Около 90 % диагнозов болезни Гентингтона, основанных на обнаружении типичных симптомов и семейном анамнезе, подтверждаются генетическим тестированием. Большинство других расстройств с аналогичными симптомами называют ХГ-подобными расстройствами (англ. HD-like disorders, HDL ) . Причины большинства HDL-заболеваний неизвестны. Известно лишь, что некоторые из них возникают в результате мутаций генов PRNP (HDL1), junctophilin 3 (HDL2), рецессивно наследуемого HTT гена (HDL3 — обнаружен у одной семьи и мало изучен) и гена, кодирующего ТАТА-связывающий белок (HDL4/SCA17) . К другим заболеваниям с аутосомно-доминантным наследованием, которые схожи с болезнью Гентингтона, относят дентаторубро-паллидолюисовую атрофию и нейроферритинопатию .
Лечение

Болезнь Гентингтона неизлечима, но существует лечение, способное облегчить некоторые симптомы .
Тетрабеназин был разработан специально для уменьшения тяжести симптомов болезни Гентингтона, был утвержден в 2008 году в США был одобрен в 2017 г. Нейролептики и бензодиазепины помогают уменьшить проявления хореи . Амантадин и находятся в стадии исследования, но показали положительные результаты . Для облегчения гипокинезии и ригидности мышц назначают противопаркинсонические лекарства, для облегчения миоклонической гиперкинезии — вальпроевую кислоту . В России препарат продается под торговым названием Нормокинезтин. С 1 января 2018 года Нормокинезтин включен в .
Для устранения депрессии применяют селективные ингибиторы обратного захвата серотонина и миртазапин , а при психозах и нарушениях поведения назначают атипичные антипсихотики .
В настоящий момент ведутся активные исследования по разработке способа лечения, исследуются потенциальные направления для лечения болезни Гентингтона . Так компания Teva исследовала препарат- иммуномодулятор , обладающий протективным действием по отношению к ЦНС . Испытания препарата дошли до II фазы, но в ходе КИ препарату не удалось достигнуть конечной точки оценки эффективности. Однако испытующие определили, что на фоне терапии лахинимодом происходит снижение скорости атрофии головного мозга . Исходя из опыта неудавшегося исследования, компания приняла решение об отказе от дальнейшего изучения лекарственного препарата .
Прогноз
С момента появления первых симптомов продолжительность жизни составляет около 15—20 лет.
Смерть обычно происходит не из-за болезни Гентингтона, а из-за сопутствующих ей осложнений, включая пневмонию, заболевания сердца и травмы. Часто больные совершают суицид .
Направление исследований
Обзор по клиническим исследованиям
На ноябрь 2021 года по всему миру было запущено 208 клинических исследований , 59 из них находятся в активном статусе .
Клинические исследования используют различные методики лечения.
Снижение уровня гентингтина
Причиной БГ является наличие мутации в одном гене, который вырабатывает токсичный для клеток вариант белка гентингтина. Одним из самых перспективных способов лечения исходит из идеи снизить уровень выработки этого варианта белка путём использования технологий подавления экспрессии генов . Исследования на мышиных моделях указывают на то, что снижение уровня мутантного белка в нервной системе улучшает их состояние . Безопасность некоторых методов подавления экспрессии генов, например, РНК-интерференции или использования (ASO), проверена в исследованиях на мышиных моделях, а также в мозге макак . У аллель-специфичного подавления имеется возможность уменьшать количество как обоих вариантов белка (дикого и мутантного), так и быть специфичным только к мутантному, например, с помощью определения однонуклеотидного полиморфизма между двумя вариантами гена . В качестве механизма подавления рассматриваются и технологии редактирования генома, например, с помощью систем на базе CRISPR/Cas9 .
По ASO уже имеются определённые результаты по нескольким исследованиям. В 2015 году компания Ionis Pharmaceuticals совместно и Институтом Нейрологии UCL запустили Фазу 1/2a клинического исследования препарата IONIS-HTTRx. Данный этап исследования должны был показать как безопасность использования на людях, так и получить первую информацию об эффективности. IONIS-HTTRx призван уменьшать экспрессию обеих копий гена. В качестве метода обнаружения снижения уровня белка гентингтина был взят метод измерения его содержания в спинномозговой жидкости. Результаты этой Фазы показали, что у всех участников, кому вводили препарат, обнаруживается статистически значимое снижения уровня гентингина. Фаза 3 была начата в 2019 году при партнёрстве с Roche Pharmaceuticals. Препарат к этому моменту был переименован на "томинерсен". Однако в марте 2021 года исследование томинерсена было остановлено регулирующими организациями из-за выявления неоптимального соотношения пользы и риска.
Имеются другие исследования, которые исследуют возможности подавления эспрессии генов. Компания Wave Life Sciences в 2021 году закончила клиническое тестирование двух препаратов, которые специфичны только для мутантного варианта гена. Результаты исследования этих препаратов оказались неудачными, на данный момент Wave Life Sciences проводит подготовку для начала тестирования третьего варианта препарата. Компания Uniqure в 2019 году анонсировала свой вариант ASO.
Примечания
Комментарии
- Джордж Хантингтон, описавший в 1872 году болезнь, названную в его честь, был не первым, кто дал её описание. Это было сделано различными авторами, как минимум пяти публикаций, начиная с 1832 года
Источники
- (англ.) — 2016.
- Monarch Disease Ontology release 2018-06-29 — 2018-06-29 — 2018.
- ↑ , с. 74.
- NCBI OMIM. . Дата обращения: 22 мая 2008. 19 ноября 2004 года.
- . Дата обращения: 29 сентября 2011. 8 марта 2021 года.
- . Дата обращения: 3 октября 2017. 19 октября 2017 года.
- ↑ Walker F.O. Huntington's disease (англ.) // The Lancet . — Elsevier , 2007. — Vol. 369 , no. 9557 . — P. 220 . — doi : . — .
- Katsuno M., Banno H., Suzuki K., et al. (англ.) // : journal. — 2008. — Vol. 8 , no. 3 . — P. 221—234 . — doi : . — . 13 февраля 2009 года.
- ↑ Walker F.O. Huntington's disease (англ.) // The Lancet . — Elsevier , 2007. — Vol. 369 , no. 9557 . — P. 221 . — doi : . — .
- Walker F.O. Huntington's disease (англ.) // The Lancet . — Elsevier , 2007. — Vol. 369 , no. 9557 . — P. 222 . — doi : . — .
- Nance M.A., Myers R.H. Juvenile onset Huntington's disease—clinical and research perspectives (англ.) // : journal. — 2001. — Vol. 7 , no. 3 . — P. 153—157 . — doi : . — .
- Vessie, P. R. (1932) «On the transmission of Huntington’s chorea for 300 years—the Bures family group». Journal of Nervous and Mental Disease, Baltimore 76: 553—573.
- Wexler, A. (2008) «The Woman Who Walked into the Sea: Huntington’s and the Making of a Genetic Disease», Yale University Press; 1 edition
- Goehler H., Lalowski M., Stelzl U., et al. (англ.) // : journal. — 2004. — Vol. 15 , no. 6 . — P. 853—865 . — doi : . — . 5 октября 2017 года.
- Harjes P., Wanker E.E. (англ.) // : journal. — 2003. — Vol. 28 , no. 8 . — P. 425—433 . — doi : . — . 5 октября 2017 года.
- ↑ Cattaneo E., Zuccato C., Tartari M. Normal huntingtin function: an alternative approach to Huntington's disease (англ.) // Nature Reviews Neuroscience : journal. — 2005. — Vol. 6 , no. 12 . — P. 919—930 . — doi : . — .
- Rubinsztein D.C., Carmichael J. Huntington's disease: Molecular basis of neurodegeneration (англ.) // Expert Rev Mol Med : journal. — 2003. — Vol. 5 , no. 20 . — P. 1—21 . — doi : . — .
- ↑ . 31 августа 2010 года.
- Arrasate, M. (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature, 431(7010):805-10.
- Subramaniam S., Sixt K.M., Barrow R., Snyder S.H. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity (англ.) // Science : journal. — 2009. — Vol. 324 , no. 5932 . — P. 1327—1330 . — doi : . — . — PMC .
- Purves D., Augustine G. A., Fitzpatrick D., Hall W., LaMantia A-S, McNamara J. O., Williams S. M. // (англ.) / Purves D.. — 2nd. — Sunderland, MA: , 2001. — ISBN 0-87893-742-0 . 18 февраля 2009 года.
- Lobsiger C.S., Cleveland D.W. Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease (англ.) // Nature Neuroscience . — 2007. — Vol. 10 , no. 11 . — P. 1355—1360 . — doi : . — .
- Crossman A.R. (англ.) // . — 2000. — Vol. 196 (Pt 4) . — P. 519—525 . — doi : . — . — PMC . (недоступная ссылка)
- ↑ Walker F. O. Huntington's disease (англ.) // The Lancet . — Elsevier , 2007. — Vol. 369 , no. 9557 . — P. 218 . — doi : . — .
- ↑ . genereviews bookshelf . University of Washington (19 июля 2007). Дата обращения: 12 марта 2009. 10 февраля 2009 года.
- Kremer B. Clinical neurology of Huntington's disease // (англ.) / Bates G., Harper P., Jones L.. — Oxford: Oxford University Press , 2002. — P. 28—53. — ISBN 0-19-851060-8 .
- Wagle A. C.; Wagle S. A., Marková I. S., Berrios G. E. Psychiatric Morbidity in Huntington's disease (англ.) // Neurology, Psychiatry and Brain Research. — 2000. — No. 8 . — P. 5—16 .
- Myers R.H. Huntington's disease genetics (англ.) // NeuroRx. — 2004. — Vol. 1 , no. 2 . — P. 255—262 . — doi : . — . — PMC .
- Kuliev A., Verlinsky Y. (англ.) // Curr. Opin. Obstet. Gynecol. : journal. — 2005. — Vol. 17 , no. 2 . — P. 179—183 . — doi : . — . 13 августа 2011 года.
- ↑ Schneider S.A., Walker R.H., Bhatia K.P. The Huntington's disease-like syndromes: what to consider in patients with a negative Huntington's disease gene test (англ.) // Nature Reviews Neurology : journal. — 2007. — Vol. 3 , no. 9 . — P. 517—525 . — doi : . — .
- Frank S., Jankovic J. (англ.) // : journal. — Adis International , 2010. — Vol. 70 , no. 5 . — P. 561—571 . — doi : . — . 8 октября 2011 года. . Дата обращения: 25 апреля 2011. Архивировано из 8 октября 2011 года.
- Walker F. O. Huntington's disease (англ.) // The Lancet . — Elsevier , 2007. — Vol. 369 , no. 9557 . — P. 224 . — doi : . — .
- . FDA Approves First Drug for Treatment of Chorea in Huntington's Disease . U.S. Food and Drug Administration (15 августа 2008). Дата обращения: 10 августа 2008. 1 июня 2012 года.
- Walker F. O. Huntington's disease (англ.) // The Lancet . — Elsevier , 2007. — Vol. 369 , no. 9557 . — P. 225 . — doi : . — .
- . genereviews bookshelf . University of Washington (19 июля 2007). Дата обращения: 12 марта 2009. 10 февраля 2009 года.
- Bonelli R. M., Wenning G. K., Kapfhammer H. P. (англ.) // : journal. — 2004. — Vol. 19 , no. 2 . — P. 51—62 . — doi : . — . 1 июня 2012 года.
- от 17 мая 2012 на Wayback Machine
- от 7 сентября 2018 на Wayback Machine remedium.ru, 06.09.2018
- от 7 сентября 2018 на Wayback Machine PharmaTimes Media, 06.09.2018
- (англ.) . ClinicalTrials.gov . Дата обращения: 11 ноября 2021. 11 ноября 2021 года.
- (англ.) . Дата обращения: 11 ноября 2021. 11 ноября 2021 года.
- Munoz-Sanjuan I, Bates GP (February 2011). . The Journal of Clinical Investigation (англ.) . 121 (2): 476—83. doi : . PMC . PMID .
- McBride JL, Pitzer MR, Boudreau RL, Dufour B, Hobbs T, Ojeda SR, Davidson BL (December 2011). . Molecular Therapy (англ.) . 19 (12): 2152—62. doi : . PMC . PMID .
- Kordasiewicz HB, Stanek LM, Wancewicz EV, Mazur C, McAlonis MM, Pytel KA, Artates JW, Weiss A, Cheng SH, Shihabuddin LS, Hung G, Bennett CF, Cleveland DW (June 2012). . Neuron (англ.) . 74 (6): 1031—44. doi : . PMC . PMID .
- Barnes DW; Whitley RJ (February 1987). . Chest (англ.) . 91 (2): 246—51. doi : . PMC . PMID .
-
Solomon, Shoshanna (2017-05-03).
.
The Times of Israel
.
из оригинала
3 мая 2017
. Дата обращения:
5 мая 2017
.
{{ cite news }}: Указан более чем один параметр|accessdate=and|access-date=( справка )
Литература
- Эдвин Кёрк. Мальчик, который не переставал расти и другие истории про гены и людей = Edwin Kirk. The genes that makes us.. — М. : Альпина нон-фикшн, 2022. — 312 с. — ISBN 978-5-00139-405-1 .
- Сара Мэннинг Пескин. В молекуле от безумия. Истории о том, как ломается мозг = Sara Manning Peskin. A Molecule Away from Madness: Tales of the Hijacked Brain. — М. : Альпина Паблишер, 2023. — С. 224. — ISBN 978-5-9614-7697-2 .
- 2020-12-03
- 1