Информационные технологии
- 1 year ago
- 0
- 0

Транскрипто́мные техноло́гии ( англ. transcriptomics technologies) — методы, разработанные для изучения транскриптома (то есть совокупности всех РНК - транскриптов ) организма. В состав транскриптома входят все транскрипты, которые присутствовали в клетке на момент . Исследуя транскриптом, можно установить, какие клеточные процессы были активны в тот или иной момент времени.
Первые попытки изучения транскриптома были предприняты в начале 1990-х годов. Благодаря развитию новых технологий в конце 1990-х транскриптомика стала важной биологической наукой. В настоящий момент в транскриптомике есть два основополагающих метода: микрочипы , позволяющие выявить наличие и количество определённых транскриптов, и секвенирование РНК (РНК-Seq), в котором используются методы секвенирования нового поколения для получения последовательностей всех транскриптов. С улучшением методик количество данных, получаемых в ходе одного транскриптомного эксперимента, увеличивалось. В связи с этим методы анализа данных также совершенствовались, чтобы обеспечить точный и эффективный анализ возрастающего объёма данных. Транскриптомные базы данных постоянно растут и становятся всё более полезными для исследователей. Это связано с тем, что правильная интерпретация данных, полученных в ходе транскриптомного эксперимента, практически невозможна без опоры на предшествующие исследования.
Измерение уровня экспрессии определённых генов в клетках разных тканей и при разных условиях или же в разные моменты времени даёт информацию о регуляторных механизмах, связанных с экспрессией генов. С помощью этих данных могут быть определены функции ранее генов. Анализ транскриптомов позволяет выявить различия в экспрессии определённых генов у разных организмов, что может быть особенно полезно для понимания молекулярных основ заболеваний человека .
Первая попытка получения части человеческого транскриптома была предпринята в 1991 году; в ходе этого исследования были получены последовательности 609 мРНК из мозга человека . В 2008 году были опубликованы два человеческих транскриптома, состоящие из миллионов последовательностей, происходящих от транскриптов 16 тысяч генов . К 2015 году были опубликованы транскриптомы сотен людей . Получение транскриптомов индивидуумов с тем или иным заболеванием, разных тканей и даже одиночных клеток в настоящее время является рутинной процедурой . Бурное развитие транскриптомики было возможно благодаря быстрому развитию новых экономичных технологий с повышенной чувствительностью .
Исследования отдельных транскриптов проводились ещё за несколько десятилетий до того, как методы транскриптомики стали общедоступны. В конце 1970-х были получены , которые были конвертированы в комплементарную ДНК ( кДНК ) с помощью обратной транскриптазы для бабочки . В 1980-х с помощью низкопроизводительного секвенирования по Сэнгеру были получены последовательности случайных транскриптов; так появились так называемые ( англ. expressed sequence tags, EST) . Метод секвенирования по Сэнгеру доминировал до появления технологий высокопроизводительного секвенирования, например, секвенирования синтезом ( Solexa/Illumina ). EST стали активно использоваться в 1990-х как эффективный метод определения генного состава организма без . Количество отдельных транскриптов оценивалось с помощью нозерн-блотов , и количественной ПЦР с обратной транскрипцией (RT-qPCR) . Однако эти методы очень трудоёмки и охватывают лишь крошечную долю целого транскриптома .
Слово «транскриптом» ( англ. transcriptome) было введено в употребление в 1990-х годах . В 1995 году появился первый транскриптомный метод, основанный на секвенировании, — ( англ. serial analysis of gene expression (SAGE)), который заключался в секвенировании по Сэнгеру соединённых фрагментов случайных транскриптов. Количество транскриптов оценивалось по числу совпадений с фрагментами известных генов . Вскоре появился вариант SAGE, использующий вместо секвенирования по Сэнгеру технологии секвенирования нового поколения — цифровой анализ экспрессии генов ( англ. digital gene expression analysis) . Однако эти методы практически полностью были вытеснены методами высокопроизводительного секвенирования целых транскриптов, которые давали дополнительную информацию о транскрипте, например, сведения о сплайсинговых вариантах .
| РНК-Seq | Микрочипы | |
|---|---|---|
| От 1 дня до 1 недели на эксперимент | 1—2 дня на эксперимент | |
| Необходимое количество РНК | Низкое ~ 1 нг тотальной РНК | Высокое ~ 1 мкг РНК |
| Трудоёмкость | Высокая ( пробоподготовка и анализ данных) | Низкая |
| Предшествующая информация | Не требуется, хотя наличие /транскриптома упрощает работу | Для создания необходим референсный геном/транскриптом |
| Точность * | ~ 90 % (ограничена последовательностей) | > 90 % (ограничена точностью детекции флуоресценции ) |
| Разрешение последовательностей | Может детектировать однонуклеотидные полиморфизмы и сплайсинговые варианты (ограничение — точность секвенирования (~ 99 %)) | Специализированные микрочипы могут детектировать сплайсинговые варианты (ограничения — создание проб и кросс-гибридизация) |
| Чувствительность | 1 транскрипт на миллион (приблизительно, ограничение — покрытие последовательности) | 1 транскрипт на тысячу (приблизительно, ограничено детекцией флуоресценции) |
| Динамический диапазон | 100000 : 1 (ограничен покрытием последовательности) | 1000 : 1 (ограничен насыщением флуоресценции) |
| Техническая воспроизводимость | > 99 % | > 99 % |
Превалирующие современные методы — микрочипы и РНК-Seq — появились в середине 1990-х и 2000-х соответственно . Публикации по микрочипам, которые измеряли относительное содержание определённых транскриптов за счёт их гибридизации с комплементарными пробами, нанесёнными на микрочип, появились в 1995 году . Метод микрочипов позволял одновременно исследовать тысячи транскриптов, и за счёт этого позволял снизить стоимость исследования транскриптома в расчёте на ген и сэкономить усилия . До конца 2000-х лучшими методами транскрипционного профилинга были пятновые олигонуклеотидные чипы ( англ. spotted oligonucleotide arrays) и микрочипы с высокой плотностью . В течение этого периода было создано множество чипов, покрывающих известные гены модельных и экономически важных организмов. Улучшения технологий создания микрочипов привели к увеличению специфичности проб и количества генов, которые можно проанализировать с помощью одного чипа. Благодаря новым методам детекции флуоресценции стало возможным точно определять наличие и количество даже транскриптов, синтезируемых на низком уровне .
РНК-Seq подразумевает секвенирование кДНК, соответствующей транскриптам, причём численность отдельных фрагментов кДНК определяется численностью соответствующих транскриптов. Огромное влияние на РНК-Seq оказало развитие методов секвенирования нового поколения . Первым транскриптомным методом стало ( англ. Massively parallel signature sequencing (MPSS)), в основе которого лежало образование коротких последовательностей длиной от 16 до 20 пар оснований (п. о.) в ходе сложной последовательности гибридизаций . В 2004 году с помощью этого метода была оценена экспрессия 10 тысяч генов растения Arabidopsis thaliana . Первая работа, посвящённая РНК-Seq, была опубликована в 2006 году. В ходе этого исследования с помощью технологии была определена последовательность ста тысяч транскриптов . Полученного покрытия было достаточно для оценки относительного количества отдельных транскриптов. Популярность РНК-Seq значительно повысилась после 2008 года, когда технологии Illumina/Solexa позволили секвенировать один миллиард транскриптов . Благодаря этим данным сейчас возможно количественно оценивать и сравнивать транскриптомы разных людей .
Получение данных о транскриптах возможно двумя принципиально различающимися путями: секвенированием отдельных транскриптов (EST или РНК-Seq) или гибридизацией транскриптов на упорядоченный чип нуклеотидных последовательностей (микрочип) .
Для всех транскриптомных методов необходимо выделить РНК из исследуемого организма. Несмотря на огромное разнообразие биологических систем, методика выделения РНК во всех случаях примерно одна и та же. Она включает разрушение клеток и тканей, разрушение РНКаз при помощи солей , разрушение макромолекул и комплексов, содержащих нуклеотиды , отделение РНК от ненужных биомолекул , включая ДНК, концентрирование РНК при помощи из раствора и очищение с помощью специальных . Выделенную РНК также можно дополнительно обработать ДНКазой , чтобы разрушить остатки ДНК . Обычно необходимо концентрирование мРНК, поскольку 98 % выделенной РНК приходится на рРНК . Концентрирование можно произвести с помощью методов, использующих наличие у мРНК поли(А)-хвоста , или путём удаления рРНК с помощью специфических проб . На результаты эксперимента может повлиять разрушенная РНК. Например, если отбирать мРНК из повреждённых РНК, то отобранные молекулы могут быть лишены 5'-концов и привести к искажению данных. Чтобы избежать разрушения РНК, перед её выделением образец обычно подвергают .
Экспрессируемые метки последовательностей (EST) — это короткие нуклеотидные последовательности, полученные из целого транскрипта. Поскольку EST можно получить без какой-либо специфики относительно организма, из которого выделена РНК, их можно получить из смеси организмов или образцов, взятых из окружающей среды . Хотя в настоящее время чаще всего используется высокопроизводительное секвенирование, EST активно использовали при разработке первых микрочипов. Например, микрочип ячменя был получен из 350 тысяч предварительно секвенированных EST .
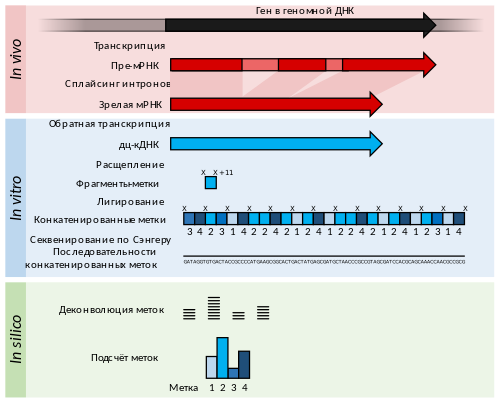
Сериальный анализ экспрессии генов является дальнейшим развитием технологии EST с большей выработкой меток. Он также позволяет провести некоторый количественный анализ численности транскриптов. РНК сначала переводится в кДНК, затем она разрезается на метки длиной 11 нуклеотидов с помощью рестриктаз, которые вносят разрывы в определённые последовательности ДНК. Полученные метки сшивают по типу «голова к хвосту» в длинные фрагменты длиной более 500 нуклеотидов, которые секвенируют с помощью низкопроизводительных, но дающих длинные методов, таких как секвенирование по Сэнгеру. Далее последовательности снова делят на 11-нуклеотидные кусочки с помощью специальных компьютерных программ (деконволюция) . Если референсный геном недоступен, то полученные метки можно непосредственно использовать в качестве диагностических маркеров, которые в случае болезни экспрессируются иначе, чем в здоровом организме .
Кэповый анализ экспрессии генов ( англ. cap analysis gene expression, CAGE) представляет собой вариант SAGE, при котором в качестве меток берутся только 5'-концевые последовательности мРНК. Поэтому, когда метки выравниваются на референсный геном, можно идентифицировать точки начала транскрипции генов. Этот метод активно используется для анализа промоторов и для клонирования полноразмерных кДНК .
SAGE и CAGE дают информацию о большем количестве генов, чем секвенирование отдельных EST, однако пробоподготовка и анализ данных в этих методах существенно сложнее .

Микрочип состоит из коротких олигонуклеотидов (проб), которые прикреплены в ячейках сетки на стеклянной подложке . Многочисленность транскриптов определяется на основании гибридизации транскриптов с этими пробами . в каждой ячейке свидетельствует о численности транскрипта, гибридизующегося с данной пробой .
Для создания микрочипа необходимо знать, хотя бы частично, геном исследуемого организма, например, в виде аннотированной последовательности или библиотеки EST; это необходимо для создания проб .
Микрочипы, использующиеся в транскриптомике, можно подразделить на два типа: пятновые чипы с низкой плотностью и чипы высокой плотности с короткими пробами . Пятновые чипы низкой плотности обычно представляют собой стеклянную основу, на которую нанесены пико литровые капли, содержащие разные фрагменты очищенной кДНК . Эти пробы длиннее, чем в чипах с короткими пробами, и с их помощью нельзя выявить события альтернативного сплайсинга . В пятновых чипах используются два типа флуорофоров , которыми метят экспериментальные и контрольные образцы, а относительная многочисленность высчитывается из интенсивности флуоресценции . Чипы высокой плотности используют только одну флуоресцентную метку, и каждый образец гибридизуется и детектируется отдельно . Чипы высокой плотности распространялись компанией Affymetrix GeneChip. В этих чипах каждому транскрипту соответствует несколько 25-нуклеотидных проб . Компания NimbleGen производит чипы высокой плотности при помощи , которая позволяла получать чипы разного строения. Один чип содержит сотни тысяч проб длиной от 45 до 85 нуклеотидов, которые гибридизуются с образцом, меченным флуоресцентной меткой одного вида .
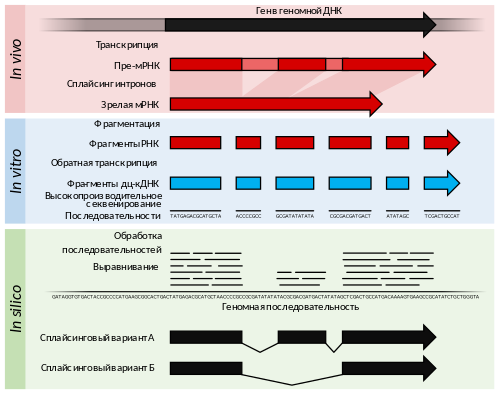
РНК-Seq представляет собой сочетание высокопроизводительного секвенирования с вычислительными методами оценки численности отдельных транскриптов в экстракте РНК . Обычно получаются последовательности длиной около 100 пар оснований (п. о.), однако в зависимости от метода секвенирования их длина может составлять от 30 п. о. до 10 тысяч п. о. РНК-Seq обеспечивает глубокое покрытие транскриптома множеством коротких фрагментов, благодаря которому возможно при помощи вычислительных методов реконструировать исходные транскрипты, выравнивая прочтения на референсный геном или друг на друга ( сборка de novo ) . С помощью РНК-Seq можно рассчитать количество как многочисленных, так и малочисленных РНК, так как динамический диапазон метода составляет 5 порядков. В этом заключается главное преимущество РНК-Seq перед микрочипами. Кроме того, для РНК-Seq требуется очень мало исследуемой РНК по сравнению с микрочипами — нанограммы против микрограммов. Благодаря этому РНК-Seq в сочетании с линейной амплификацией кДНК позволяет исследовать очень небольшие клеточные структуры вплоть до отдельных клеток . Теоретически верхнего предела количественной оценки в РНК-Seq не существует, и для прочтений длиной 100 п. о. фоновый шум в неповторяющихся участках очень низок .
С помощью РНК-Seq можно идентифицировать гены в геноме или установить, какие гены активны в данный момент времени. На основании количества прочтений можно точно установить относительный уровень экспрессии генов. Методология РНК-Seq постоянно совершенствуется, преимущественно за счёт улучшения технологий секвенирования, которые повышают производительность и точность метода, а также выдают прочтения всё большей длины . Со времени первых публикаций в 2006 и 2008 годах РНК-Seq интенсивно внедрялся в исследования и к 2015 году догнал микрочипы, став вторым доминирующим транскриптомным методом .
Попытки получения транскриптомных данных для отдельных клеток стимулировали совершенствование методов приготовления библиотек для РНК-Seq, что значительно увеличило чувствительность технологии. На данный момент получен ряд транскриптомов единичных клеток и даже появились методы РНК-Seq in situ , в которых транскриптомы отдельных клеток были получены непосредственно в фиксированных тканях .
РНК-Seq появился вместе с бурным развитием нескольких методов высокопроизводительного секвенирования . Однако стадии секвенирования выделенных РНК предшествуют несколько этапов пробоподготовки, которые различаются в разных методах. Методы различаются способами концентрирования транскриптов, фрагментации, амплификации, способом секвенирования (одноконцевое или парноконцевое), а также тем, сохраняется ли информация об исходной цепи .
Чувствительность РНК-Seq в конкретном эксперименте можно повысить за счёт концентрирования интересующих классов РНК и удаления остальных. мРНК можно отделить с помощью олигонуклеотидных проб, которые связываются с их поли(А)-хвостами. Удалить неинформативные и чрезвычайно многочисленные рРНК можно с помощью гибридизующихся проб, созданных специально для рРНК данного таксона (например, млекопитающих или растений). Однако вместе с рРНК с помощью такого подхода можно удалить и другие РНК, что может исказить картину эксперимента. Малые РНК , например, микроРНК , можно выделить на основании их размера из агарозного геля после .
Поскольку мРНК, как правило, длиннее, чем единичные прочтения в большинстве методов высокопроизводительного секвенирования, обычно перед секвенированием транскрипты . Метод фрагментации лежит в основе создания библиотеки для секвенирования. Фрагментацию можно проводить путём химического гидролиза , распыления, обработки ультразвуком ( ) или обратной транскрипции с использованием терминирующих нуклеотидов . Кроме того, фрагментацию и добавление меток к кДНК можно осуществить одновременно при помощи транспозаз .
В ходе пробоподготовки для секвенирования фрагменты кДНК, соответствующие транскриптам, можно размножить с помощью ПЦР так, чтобы повысить численность молекул, содержащих необходимые 3'- и 5'-концевые . Стадия амплификации также необходима перед секвенированием образцов с очень низким содержанием РНК. Нижним пределом количества РНК, которое пригодно для секвенирования, является 50 пикограмм . Чтобы оценить качество библиотеки и секвенирования ( GC-состав , длина фрагментов, предпочтение фрагментов с определённым положением в транскрипте), можно использовать контрольные . ( англ. unique molecular identifiers, UMI) — это короткие случайные последовательности, которые используются для индивидуального мечения фрагментов при приготовлении библиотеки таким образом, чтобы после добавления идентификатора каждый фрагмент был уникален . С помощью UMI можно измерить численность транскриптов в абсолютной шкале, чтобы скорректировать отклонения, возникшие при создании библиотеки, перед амплификацией, а также чтобы точно оценить количество ДНК в исходном образце. UMI особенно удобны для РНК-Seq одиночных клеток, в которых исходное количество РНК очень мало и требует неспецифичной амплификации .
После пробоподготовки молекулы транскриптов (точнее, соответствующих им кДНК) можно секвенировать в одном направлении (одноконцевое прочтение) или в обоих (парноконцевое прочтение). Одноконцевое секвенирование, как правило, быстрее и дешевле, и в большинстве случаев его достаточно для количественной оценки уровней экспрессии генов. Парноконцевое секвенирование позволяет получить более точные выравнивания и сборки , что очень важно для аннотации генов и описания изоформ транскриптов . Методы РНК-Seq, специфичные по отношению к цепи, сохраняют сведения о цепи ДНК, с которой был транскрибирован каждый транскрипт. Без этой информации прочтения можно выровнять на локус , однако будет неясно, в каком направлении происходит транскрипция гена. Одноцепочечный РНК-Seq удобен для определения направления транскрипции , расположенных на разных цепях, что позволяет сделать предсказание генов у немодельных организмов более точным .
| Платформа | Коммерческий релиз | Типичная длина прочтения | Максимальная производительность одного запуска | Точность единичного прочтения | Запуски РНК-Seq, размещённые в базе NCBI SRA по состоянию на октябрь 2016 года. RNA-Seq runs deposited in the NCBI SRA (Oct 2016) |
|---|---|---|---|---|---|
| 454 Life Sciences | 2005 | 700 п. о. | 0,7 миллиарда п. о. | 99,9 % | 3548 |
| Illumina | 2006 | 50-300 п. о. | 900 миллиардов п. о. | 99,9 % | 362903 |
| SOLiD | 2008 | 50 п. о. | 320 миллиардов п. о. | 99,9 % | 7032 |
| Ion Torrent | 2010 | 400 п. о. | 30 миллиардов п. о. | 98 % | 1953 |
| 2011 | 10000 п. о. | 2 миллиарда п. о. | 87 % | 160 |
NCBI SRA — Национального центра биотехнологической информации ( США )
Поскольку в настоящий момент РНК-Seq включает перевод РНК в кДНК в ходе обратной транскрипции, платформы для последующего секвенирования одинаковы и для транскриптомных, и для геномных данных. По этой причине развитие РНК-Seq в значительной мере определяется усовершенствованием методов секвенирования ДНК . Однако всё большее распространение получает непосредственное секвенирование РНК при помощи нанопор . С помощью нанопорового секвенирования в РНК можно обнаружить модифицированные основания , которые нельзя было выявить при секвенировании кДНК, кроме того, для этого метода не нужна амплификация, которая вносит дополнительные искажения .
Чувствительность и точность РНК-Seq определяются числом прочтений, полученных с каждого образца . Для достаточного покрытия транскриптома необходимо очень много прочтений, что позволяет обнаружить даже малочисленные транскрипты. Дополнительные сложности создаёт этап секвенирования, дающего прочтения ограниченной длины, различной точности и качества. Более того, организмы каждого вида имеют разное количество генов, поэтому для эффективной сборки транскриптома для каждого вида необходимо разное число прочтений. На ранних этапах это количество определялось эмпирически, однако с развитием технологий необходимое покрытие стало возможным предсказывать вычислительными методами. Наиболее эффективным способом улучшения точности выявления дифференциальной экспрессии низкоэкспрессируемых генов является не увеличение числа прочтений, а увеличение . В настоящий момент Энциклопедия элементов ДНК рекомендует 70-кратное покрытие экзома для обычного РНК-Seq и до 500-кратного покрытия для обнаружения редких транскриптов и изоформ .
Методы транскриптомики позволяют проводить параллельные эксперименты со множеством образцов, поэтому для получения результатов как с помощью РНК-Seq, так и с помощью микрочипов необходима серьёзная вычислительными методами . Данные с микрочипов представляют собой изображения с высоким разрешением , поэтому обработка данных включает ( англ. feature detection) и спектральный анализ . Изображения, полученные с микрочипов, достигают размера 750 Мб , в то время как обработанные данные занимают 60 Мб. Множество коротких проб, соответствующих одному и тому же транскрипту, могут позволить определить экзон - интронную структуру гена, поэтому для определения достоверности финального сигнала необходимы статистические модели . В ходе экспериментов РНК-Seq получаются миллиарды коротких последовательностей ДНК, которые необходимо выровнять на референсный геном , включающий миллионы или миллиарды п. о. Сборка транскриптома de novo требует построения весьма сложных . Операции обработки данных РНК-Seq требуют многократного повторения, поэтому для них могут удобны параллелизованные вычисления , однако с использованием современных алгоритмов обработку данных простых транскриптомных экспериментов, не требующих сборки de novo , можно осуществить даже на обычном персональном компьютере . Человеческий транскриптом можно достаточно точно собрать из 300 миллионов 100-нуклеотидных прочтений, полученных с помощью РНК-Seq . Для хранения такого объёма данных в сжатом требуется 1,8 Гб дискового пространства на образец. Обработанные численные значения для каждого гена занимают ещё меньше памяти, сопоставимо с обработанными данными с микрочипов. Данные о последовательностях можно хранить в публичных данных, таких как SRA (от англ. sequence read archive — архив прочтений последовательностей) . Набор данных РНК-Seq можно загрузить в базу данных Gene Expression Omnibus .
Обработка изображений , полученных с микрочипов, должна сохранять ячеек изображения и независимо количественно оценивать интенсивность флуоресценции в каждой ячейке. Необходимо также выявлять и исключать их из финального анализа. Интенсивность флуоресценции свидетельствует о представленности каждой последовательности, поскольку последовательность пробы в каждой ячейке известна .
Первые этапы РНК-Seq также включают схожую обработку изображений, однако перевод изображений в данные о последовательностях производится автоматически специальными программами. Результатом секвенирования синтезом по технологии Illumina является совокупность кластеров, расположенных на поверхности проточной ячейки . В ходе каждого цикла секвенирования изображение каждой проточной ячейки снимается до четырёх раз, причём один запуск включает десятки или сотни циклов. Кластеры проточных ячеек аналогичны пятнам в микрочипах и на ранних этапах секвенирования должны быть правильно определены. При пиросеквенировании (компания ) интенсивность испускаемого света соответствует количеству одинаковых нуклеотидов в гомо полимерном участке. Существует множество вариаций перечисленных методов, и каждая предполагает использование разных профилей ошибок для получающихся данных .
В ходе экспериментов РНК-Seq получается огромный объём прочтений, которые необходимо обработать для получения полезной информации. Анализ данных, как правило, включает использование комбинаций различных биоинформатических программ, которые необходимо подбирать сообразно эксперименту и целям. Процесс обработки данных можно разбить на четыре этапа: контроль качества, выравнивание, количественный анализ и дифференциальная экспрессия . Наиболее популярные программы для обработки данных РНК-Seq запускаются из командной строки в среде Unix или R / Bioconductor .
Прочтения небезупречны, поэтому необходимо определить точность прочтения каждого основания в последовательности. Прочтения, прошедшие контроль качества, гарантированно имеют высокую точность определения каждого основания, их GC-состав соответствует ожидаемому распределению, в них нет перепредставленности коротких мотивов и редки дупликации . Существует несколько программ для анализа качества, например, FastQC и FaQCs. Низкокачественные прочтения или удаляются, или специальным образом помечаются, что учитывается при дальнейшем анализе .
Чтобы связать численность прочтений с определённым геном, прочтения необходимо выровнять на референсный геном или друг на друга, если референсный геном неизвестен (сборка транскриптома de novo ) . Основными требованиями, которым должны удовлетворять программы для выравнивания, являются скорость, позволяющая за приемлемое время выровнять миллиарды коротких прочтений, определённая гибкость, чтобы выявлять случаи сплайсинга эукариотических мРНК, а также правильно выбирать расположение прочтений, соответствующих нескольким местам в геноме. Программы постоянно совершенствуются в соответствии с перечисленными требованиями, а увеличение длины прочтений снижает вероятность неоднозначного выравнивания. (EBI) поддерживает список доступных в данный момент инструментов для выравнивания прочтений, полученных при высокопроизводительном секвенировании .
Выравнивание первичных эукариотических транскриптов на референсный геном требует особое обращение с интронами, которых нет в зрелых мРНК . Программы для выравнивания коротких прочтений могут создавать особые выравнивания, предназначенные специально для идентификации сайтов сплайсинга на основе канонических последовательностей сайтов сплайсинга. Идентификация сайтов сплайсинга предотвращает их ошибочное выравнивание или отбраковывание, позволяя выровнять на референсный геном большее количество прочтений и увеличивая качество количественных оценок экспрессии генов. Поскольку * может осуществляться на уровне изоформ мРНК, выравнивания, учитывающие сплайсинг, позволяют обнаружить изменения в численности тех или иных изоформ, что было бы невозможно с помощью обычного анализа .
Для сборки транскриптома de novo производится выравнивание прочтений друг на друга, что позволяет реконструировать полноразмерные транскрипты без использования референсного генома . Сложностями сборки de novo являются необходимость больших вычислительных мощностей, чем для сборки на основе референсного генома, дополнительной проверки вариантов и фрагментов генов и дополнительной аннотации собранных транскриптов. Первые метрики, предназначенные для оценки качества сборки транскриптома, такие как N50 , были признаны ошибочными , и в данный момент доступны усовершенствованные методы оценки. Метрики, основанные на аннотации, хорошо подходят для оценки степени сборки генома. Транскриптом, собранный de novo , может быть использован в качестве референсного при выравнивании последовательностей и количественном анализе экспрессии генов .
| Программа | Дата релиза | Дата последнего обновления | Вычислительная эффективность | Сильные и слабые стороны |
|---|---|---|---|---|
| Velvet-Oases | 2008 | 2011 | Низкая, один поток выполнения , необходимо много памяти с произвольным доступом | Первый сборщик коротких прочтений. В настоящее время почти не используется. |
| SOAPdenovo-trans | 2011 | 2014 | Средняя, несколько потоков выполнения, умеренная необходимость в памяти с произвольным доступом | Один из первых сборщиков коротких прочтений. Адаптирован для сборки транскриптомов. |
| Trans-ABySS | 2010 | 2016 | Средняя, несколько потоков выполнения, умеренная необходимость в памяти с произвольным доступом | Предназначена для коротких прочтений, но может быть использована и для сложных транскриптомов. Доступна MPI -параллельная версия для вычислительных кластеров. |
| Trinity | 2011 | 2017 | Средняя, несколько потоков выполнения, умеренная необходимость в памяти с произвольным доступом | Предназначена для коротких прочтений. Можно использовать для сложных транскриптомов, но требуется много памяти. |
| miraEST | 1999 | 2016 | Средняя, несколько потоков выполнения, умеренная необходимость в памяти с произвольным доступом | Может обрабатывать повторяющиеся последовательности, комбинирует несколько форматов данных секвенирования, совместима с большим числом платформ секвенирования. |
| Newbler | 2004 | 2012 | Низкая, один поток выполнения, необходимо много памяти с произвольным доступом | Специализирована на устранении ошибок секвенаторов 454 Roche, связанных с гомополимерными последовательностями. |
| CLC genomics workbench | 2008 | 2014 | Высокая, несколько потоков выполнения, низкая необходимость памяти с произвольным доступом | Имеет графический интерфейс , может комбинировать различные технологии секвенирования. Не специализирована для транскриптомов, перед использованием необходимо приобрести лицензию. |
| SPAdes | 2012 | 2017 | Высокая, несколько потоков выполнения, низкая необходимость памяти с произвольным доступом | Предназначена для транскриптомных экспериментов с единичными клетками. |
| RSEM | 2011 | 2017 | Высокая, несколько потоков выполнения, низкая необходимость памяти с произвольным доступом | Может оценивать частоту альтернативно сплайсированных транскриптов. Удобна в использовании. |
| StringTie | 2015 | 2018 | Высокая, несколько потоков выполнения, низкая необходимость памяти с произвольным доступом | Может использовать комбинацию методов сборки на основе референсного генома и de novo для идентификации транскриптов. |
Количественный анализ выравниваний прочтений может быть произведён на уровне гена, экзона и транскрипта. Типичным результатом анализа является количество прочтений для каждого элемента анализа (гена, экзона или транскрипта). Например, для генов она выдаётся в формате general feature format (GFF) . Количество прочтений для генов и экзонов можно определить при помощи разных программ, например, HTSeq . Анализ на уровне транскрипта более сложен и требует привлечения вероятностных методов для оценки численности транскрипта на основании коротких прочтений; например, это может выполнить программа cufflinks . Прочтения, одинаково хорошо ложащиеся на разные места в геноме, должны быть идентифицированы и удалены либо выровнены на одно из возможных мест либо на наиболее вероятное из них. Некоторые методы оценки вовсе не предполагают выравнивания прочтения на референсный геном. Например, метод, используемый в программе kallisto, объединяет псевдовыравнивание и количественный анализ в один шаг, который идёт на два порядка быстрее, чем методы программ tophat и cufflinks, и требует меньших вычислительных затрат .
Когда для каждого транскрипта получены количественные данные, с помощью их статистического анализа , моделирования и нормализации анализируется дифференциальная экспрессия генов . Большинство программ, которые её анализируют, берут на вход таблицу из названий генов и числа транскриптов для каждого из них, но некоторые программы, например, cuffdiff, в качестве входных данных получают выравнивание прочтений в (от англ. Binary Alignment Map — карта попарных выравниваний). На выходе программы выдают список генов с результатами попарных статистических тестов , проверяющих значимость различий в экспрессии между экспериментальными и контрольными данными .
| Программа | Среда | Специализация |
|---|---|---|
| Cuffdiff2 | Основанные на Unix | Анализ транскриптов, направленный на обнаружение событий альтернативного сплайсинга мРНК |
| EdgeR | R/Bioconductor | Любые количественные геномные данные |
| DEseq2 | R/Bioconductor | Разные типы данных |
| Limma/Voom | R/Bioconductor | Microarray or RNA-Seq data, flexible experiment design |
| Ballgown | R/Bioconductor | Эффективное и чувствительное отыскание транскриптов |
Результаты транскриптомного анализа можно подтвердить с помощью других методов, например, количественной ПЦР (qPCR) . Экспрессия генов измеряется относительно стандартной экспрессии изучаемого гена и контрольных генов. Принцип измерения в qPCR такой же, что и в РНК-Seq, а именно, значение для данного гена рассчитывается на основе концентрации участка-мишени в исследуемом образце. Однако qPCR подходит только для , имеющих менее 300 п. о. и расположенных вблизи 3'-конца кодирующей области . Если необходимо проверить данные по изоформам транскрипта, с помощью тщательного анализа выравниваний прочтений РНК-Seq можно определить, каким участкам должны соответствовать праймеры для qPCR, чтобы сделать различия наиболее явными . Измерение экспрессии контрольных генов наряду с исследуемыми даёт стабильные референсные данные. Проверка данных РНК-Seq с помощью контрольной ПЦР показала, что разные варианты РНК-Seq в целом дают схожие данные .
Для анализа транскриптомных данных очень важна информация о функциях изучаемых генов. Наблюдаемые паттерны экспрессии генов можно связать с определённым фенотипом при помощи экспериментов по нокдауну изучаемых генов и .
Транскриптомные технологии нашли применение в различных областях биомедицины , в частности, в диагностике и профилировании заболеваний . С помощью РНК-Seq стало возможным обнаружение сайтов начала транскрипции, использования альтернативных промоторов и новых вариантов альтернативного сплайсинга. Поскольку геномные играют важную роль в патогенезе многих заболеваний, определение их вариантов чрезвычайно важно для интерпретации данных полногеномного поиска ассоциаций . С помощью РНК-Seq можно обнаруживать однонуклеотидные полиморфизмы, связанные с болезнями, случаи аллелеспецифичной экспрессии, , которые позволяют пролить свет на генетические основы развития заболеваний .
С помощью РНК-Seq можно получить информацию о транскрипции эндогенных ретротранспозонов , которые могут влиять на транскрипцию соседних генов посредством разнообразных эпигенетических механизмов, что может привести к развитию болезней . Важной потенциальной сферой применения РНК-Seq является исследование молекулярных основ нарушений иммунной системы , потому что этот метод позволяет разделить популяции иммунных клеток разных типов и секвенировать репертуары T- и B-клеточных рецепторов пациентов .
С помощью РНК-Seq можно находить изменения в экспрессии генов у человеческих патогенов , что может помочь идентифицировать новые , предсказывать устойчивость к антибиотикам , а также понять детали . С помощью РНК-Seq можно разработать оптимизированные меры , а также направленные индивидуальные стратегии лечения .
Транскриптомный анализ можно проводить и для хозяина, и для патогена. С помощью двойного РНК-Seq можно одновременно построить профили экспрессии генов и хозяина, и патогена в ходе всего инфекционного процесса . Такой подход позволяет изучать динамический иммунный ответ и межвидовые для обоих взаимодействующих организмов с момента начального контакта до инвазии и финальной персистенции патогена или его разрушения иммунной системой хозяина .
Транскриптомика позволяет идентифицировать гены и метаболические пути , ответственные за ответ и противодействие стрессам, связанным с биотическими и абиотическими факторами внешней среды . Благодаря неспецифичным методам транскриптомики с её помощью можно находить новые генные сети даже в сложных системах. Например, сравнительный анализ нескольких линий нута на различных стадиях развития позволил идентифицировать транскрипционные профили, связанные со стрессами, вызванными засухой и повышенной солёностью; в частности, была показана роль изоформ транскриптов - . Изучение экспрессии генов при формировании биоплёнок патогенными дрожжами Candida albicans позволило выявить набор совместно регулируемых генов, критически важных для образования и поддержания биоплёнки .
Транскриптомное профилирование даёт ценнейшую информацию о механизмах лекарственной устойчивости . Анализ более тысячи малярийного плазмодия Plasmodium falciparum показал, что устойчивость к артемизинину изолятов из Юго-Восточной Азии связана с повышенной активностью и с более медленным прохождением внутри эритроцитарной стадии жизненного цикла .
Одно из применений транскриптомных технологий заключается в определении функций генов, а также аллелей, ответственных за конкретный фенотип. Транскриптомика экотипов растения Arabidopsis , которые металлы , показала связь с этим фенотипом генов, отвечающих за проникновение металлов в организм, толерантность и гомеостаз . Объединение данных РНК-Seq, полученных для разных тканей, позволило усовершенствовать аннотацию функций генов у коммерчески важных организмов, например, огурца , или видов, находящихся под угрозой исчезновения, таких как коала .
Сборка прочтений РНК-Seq не зависит от референсного генома , поэтому этот метод идеально подходит для изучения экспрессии генов у немодельных организмов, для которых ещё нет готовых геномных данных. Например, база данных однонуклеотидных полиморфизмов, которая использовалась в программах по размножению псевдотсуги Мензиса , была создана при транскриптомном анализе de novo при отсутствии секвенированного генома . Схожим образом гены, участвующие в развитии сердечной , мышечной и нервной тканей у омара , были идентифицированы путём сравнения транскриптомов разных тканей без использования последовательности генома. РНК-Seq также можно использовать для обнаружения ранее неизвестных белоккодирующих областей в уже секвенированных геномах .
Обычно транскриптомика рассматривает только мРНК клетки. Однако те же методы можно применить и к некодирующим РНК , которые участвуют в трансляции , репликации геномной ДНК , сплайсинге и регуляции транскрипции . Многие из этих некодирующих тРНК связаны с развитием болезней, в том числе рака , сердечно-сосудистых заболеваний и .
При изучении транскриптомов создаются огромные объёмы данных, которые потенциально могут быть использованы и в других проектах. Поэтому сырые или обработанные данные размещают в публичных базах данных, чтобы сделать их доступными для всего научного сообщества. Например, по состоянию на 2018 год, база данных Gene Expression Omnibus содержит данные миллионов экспериментов .
| Название | Владелец | Данные | Описание |
|---|---|---|---|
| Gene Expression Omnibus | NCBI | Микрочипы, РНК-Seq | Первая база данных транскриптомов, полученных из разных источников. Первая ввела стандарты и MINSEQE, которые регламентируют необходимые метаданные для эксперимента, чтобы он был хорошо интерпретируем и воспроизводим . |
| ArrayExpress | Микрочипы | Импортирует наборы данных из Gene Expression Omnibus и подчиняется ей. Обработанные данные и метаданные экспериментов хранятся в ArrayExpress, а сырые прочтения — в ENA. Соответствует стандартам MIAME и MINSEQE . | |
| EBI | Микрочипы, РНК-Seq | Содержит данные о тканеспецифичной экспрессии генов у животных и растений. Содержит данные вторичных анализов и их визуализацию, использует термины Gene Ontology , домены и метаболические пути. Содержит ссылки на данные по многочисленности белков, если они имеются. | |
| Частное курирование | Микрочипы, РНК-Seq | Содержит справочные пояснения к публично доступным транскриптомным данным, в основном касающиеся медицины и биологии растений. Данные отдельных экспериментов нормализованы, что позволяет сравнивать экспрессию генов в различных экспериментах. Для полного доступа необходимо приобрести лицензию, бесплатно доступна лишь часть базы. | |
| RefEx | DDBJ | Все | Транскриптомы, полученные из 40 различных органов человека, мыши и крысы . Данные по экспрессии генов визуализированы в виде тепловой карты , наложенной на трёхмерную модель анатомической структуры. |
| NONCODE | noncode.org | РНК-Seq | Некодирующие РНК (кроме тРНК и рРНК) |