Interested Article - ChIP-seq

- 2020-12-14
- 1
ChIP-seq — метод анализа ДНК - белковых взаимодействий, основанный на иммунопреципитации хроматина (ChIP) и высокоэффективном секвенировании ДНК. Метод был разработан для изучения модификаций гистонов по всему геному , а также поиска мест связывания транскрипционных факторов . Ранее самым популярным методом для установления ДНК-белковых взаимодействий был , сочетающий иммунопреципитацию хроматина с гибридизацией на ДНК-микрочипах .
Методика

Иммунопреципитация хроматина (ChIP)
Иммунопреципитация хроматина — метод, используемый для специфического накопления коротких последовательностей ДНК, связанных с исследуемым белком в живых клетках . Типичная методика включает в себя следующие стадии :
- образование обратимых сшивок между ДНК и взаимодействующими с ней белками (обычно с помощью обработки формальдегидом)
- выделение ДНК и расщепление на фрагменты ультразвуком или эндонуклеазами
- осаждение специфическими к исследуемому белку антителами , пришитыми к белкам A / G на поверхности магнитных
- разрушение сшивок между белком и ДНК, очистка ДНК
В результате выделится вся ДНК, но образец будет обогащён фрагментами, с которыми был связан исследуемый белок .
Секвенирование
Данная стадия включает в себя определение первичной последовательности полученных после иммунопреципитации фрагментов ДНК любым доступным способом. В отличие от ChIP-on-Chip, в ChIP-seq для определения последовательности ДНК используется секвенирование нового поколения . В ChIP-seq чаще используется одноконцевое секвенирование, однако использование парноконцевого секвенирования повышает точность картирования (что особенно важно для картирования повторов ) . В результате получается набор коротких перекрывающихся последовательностей (чтений, или ридов). Обычно исходные фрагменты ДНК имеют длину 150—500 п .н. , а полученные риды чаще всего имеют длину 50 п. н.
Биоинформатический анализ
Биоинформатический анализ включает в себя следующие стадии :
- Фильтрация чтений с низким качеством
- Для фильтрации полученных ридов можно использовать программные пакеты и . Определение качества чтений базируется на — весе, который присваивается каждому нуклеотиду при его прочтении. Для оценки и улучшения качества ридов могут быть использованы такие программные пакеты, как , , и Cutadapt. Gencore удаляет повторяющиеся риды, оставляя один консенсусный. Благодаря этому на выходе получаются более чистые данные чем в случае простого удаления дубликатов. Picard — набор инструментов позволяющий работать с альтернативными форматами: SAM / BAM / CRAM и VCF. FQStat — автономный, независимый от платформы программный пакет инструментом, который оценивает качество файлов FASTQ с помощью параллельного программирования. Помимо этого Illumina предоставлят внутреннюю услугу по проверке качества ридов Illumina chastity filter.
- Также для улучшения качества чтений может быть полезен «тримминг» — обрезание концов чтений с низким качеством, получающихся из-за рассогласованности (особенность секвенирования нового поколения). Тримминг производится с помощью программы Trimmomatic .
- Картирование чтений на геном
- Картирование представляет собой определение того, какой именно участок и какой хромосомы был прочитан данным конкретным чтением. Для картирования чтений на геном могут использоваться такие программные пакеты, как , , Bowtie 2 и .
- Фильтрация артефактов и чтений, откартированных в несколько мест
- Чтения, получаемые в результате массивного параллельного секвенирования, обычно имеют небольшую длину (100—200 нуклеотидов ), тогда как в средней эукариотической хромосоме порядка 100 миллионов нуклеотидов. Картирование чтений на геном не всегда представляет собой тривиальную задачу из-за наличия в геноме эукариот большого числа повторов (например, и SINE — повторы, составляющие соответственно 17 % и 11 % от последовательности ДНК человека), и, таким образом, чтения повторов могут картироваться сразу в нескольких местах. Обычно для анализа (например, транскрипционных факторов ) достаточно уникально откартированных ридов, однако в некоторых случаях в анализ включают и риды, картированные на несколько участков . В качестве альтернативы для поправки на потерянный в плохо картируемых областях сигнал может использоваться картируемость — показатель, зависящий от различных параметров эксперимента и анализа, в том числе от длины ридов и программ, используемых для обработки данных .
- Для фильтрации может быть использован программный пакет .
- Определение качества картирования
- После картирования становится возможным определить участки связывания исследуемого белка в геноме по количеству картированных на данный участок чтений (если много — белок там был) .
- Набор полученных в результате иммунопреципитации чтений может оказаться неудачным для дальнейшего анализа из-за недостаточной глубины секвенирования, неудачного выбора размера фрагментов, на которые расщеплялась ДНК при иммунопреципитации, или же недостаточной представленности связанных с исследуемым белком фрагментов в полученной после иммунопреципитации смеси (плохие антитела и т. п.).
- Для определения всего вышеперечисленного используется программный пакет .
- Выявление сайтов связывания
- После картирования ридов на геном для выявления сайтов (участков) связывания вначале оценивается уровень покрытия. Далее выявляются пики (участки с большим покрытием, где, вероятно, был связан исследуемый белок), отделяется шум и определяются границы пиков. При этом важно соблюдение баланса между чувствительностью и специфичностью .
- Некоторые из программных пакетов, которые можно использовать для решения этой задачи — , PeakSeq , , MACS 2, UGENE . Результатом работы этих программ является список участков, отранжированный либо по величине абсолютного сигнала (то есть числу ридов), либо по значимости обогащения (например, по p-value или). Выбор подходящего метода зависит от изучаемого вида и белка и условий эксперимента. Разные программы используют разные предположения и допущения для вычисления p-value и FDR. Например, SPP и исходная версия MACS используют только данные эксперимента ChIP-Seq и контроля (при его наличии), в то время как MOSAiCS учитывает показатель картируемости и GC-состав . Поэтому сравнивать результаты работы разных алгоритмов вызова пиков достаточно затруднительно. Многие работы по сопоставлению алгоритмов используют валидацию числа найденных пиков с помощью данных экспериментов ChIP-on-Chip, qPCR и т. д. . Ситуация также осложняется плохой аннотированностью истинных сайтов связывания, поэтому при поиске пиков для белка с неизвестным сайтом связывания необходимо использовать отрицательные контроли .
- Аннотация сайтов связывания
- Целью аннотации является установление связи между сайтом связывания и функциональным участком ДНК, на который попал сайт связывания. Таким функциональным участком может быть промотор , участок начала транскрипции , межгенный участок и т. п. .
- Пересечение предсказанных участков связывания с функциональными элементами ДНК может быть визуально проанализировано в одном из ; можно также получить текстовый файл с аннотацией с помощью , или .
- Выявление мотивов
- В полученных пиках (длина порядка сотен нуклеотидов) иногда можно выявить характерные последовательности, по которым происходит связывание белка — мотивы (длина обычно около 20 нуклеотидов). Для поиска мотивов можно использовать алгоритм MEME , , . Если же для исследуемого белка уже известен мотив, по которому происходит связывание, то его наличие в пиках может служить хорошим индикатором качества ChIP-seq .
Характеристики метода
При дизайне эксперимента ChIP-seq и дальнейшем биоинформатическом анализе необходимо учитывать некоторые факторы и ограничения методики :
Неравномерная фрагментация и контроль
Доступность хроматина при фрагментации не одинакова в разных частях генома: в активно транскрибируемых областях он доступнее, поэтому соответствующие фрагменты ДНК будет преобладать в образце, что может привести к ложно-положительному результату. Плотно упакованные участки, напротив, могут хуже подвергаться фрагментации и, следовательно, будут менее представлены в образце, что может привести к ложно-отрицательному результату .
Из-за неравномерной фрагментации и других факторов важно использовать правильный контроль. Консорциум ENCODE описывает два основных типа контролей . В первом варианте в качестве контроля используется ДНК, выделенная из клеток в тех же условиях, но без преципитации (так называемый контроль входной («input») ДНК). Во втором типе проводится ещё один эксперимент ChIP с использованием антител, которые связывают незначимые внеядерные антигены (так называемый «IgG контроль»). В обоих случаях глубина секвенирования должна быть не меньше глубины эксперимента ChIP-seq .
Количество клеток
У классической методики существует ряд ограничений. Так, обычно для ChIP необходимо значительное количество клеток (около 10 миллионов), что затрудняет применение данного метода на маленьких модельных организмах , а также ограничивает количество экспериментов, которые можно провести с ценным образцом. Для преодоления данного ограничения был разработан ряд методов, основанных на амплификации ДНК после ChIP-seq (например, nano-ChIP-seq). ChIP-seq отдельных клеток ( англ. Single-cell ChIP-seq) очень сложен из-за фонового шума, вызванного неспецифическим связыванием антител, и к середине второго десятилетия XXI века была опубликована лишь одна работа, в которой Single-cell ChIP-seq осуществлён успешно. В этом исследовании использовали капельную микрофлюидику, и из-за низкого покрытия потребовалось отсеквенировать тысячи клеток, чтобы выявить клеточную гетерогенность .
Отношение сигнал/шум
Отношение сигнал/шум (S/N) определяется числом и мощностью пиков, полученных для каждого образца, и может быть использовано для оценки уровня шума. Высокое значение S/N не гарантирует правильность определения сайтов связывания, а всего лишь отражает наличие большого количества участков генома, на которые откартировалось много ридов . Для определения этого показателя ENCODE предлагает две метрики :
- доля ридов в пиках (fraction of reads in peaks, FRiP) положительно коррелирует с числом и интенсивностью пиков и определяется как FRiP = ¼ N peak /N nonred , где N peak — число ридов, откартированных в районы пиков. К минусам этого показателя относится тот факт, что он зависит от глубины секвенирования и параметров поиска пиков, но в целом он положительно коррелирует с числом найденных пиков;
- кросс-корреляционные профили (cross-correlation profiles, CCPs) используют кластеризацию ридов на основании кросс-корреляции Пирсона без предварительного поиска пиков, что отличает этот метод от FRiP.
Глубина секвенирования
Глубина секвенирования (покрытие) — число уникальных ридов, откартированных на данный участок референсного генома. Глубина секвенирования влияет на выявление пиков: их число растёт с увеличением глубины секвенирования, так как с ростом числа ридов большее количество сайтов становится статистически значимым . Поэтому для распознавания всех функциональных сайтов необходимо глубокое секвенирование .
Значение достаточного уровня покрытия зависит от отношения сигнал/шум антитела и может быть определено как глубина секвенирования, при которой отношение числа пиков из случайно взятой подвыборки ридов к числу пиков из полной выборки ридов выходит на плато. Такое насыщение может быть достигнуто не всегда (например, его нет для гистонов ), и в таких случаях эта величина задаётся эмпирически .
Сложность библиотеки
Сложность библиотеки (NRF) определяется как отношение числа необогащенных ридов N nonred к общему числу откартированных ридов N all . Необогащенные риды определяются как риды, откартированные на один и тот же участок генома T раз и меньше (значение T задаётся в качестве параметра). Обогащённые риды (риды, не вошедшие в N nonred ) не рассматриваются в дальнейшем анализе. Для человека параметр T обычно берут равным 1, так как ожидаемая глубина секвенирования в этом случае обычно намного меньше единицы. Для маленьких геномов глубина секвенирования может быть больше 1, поэтому стоит взять большее значение T. При сравнении показателя NRF для разных образцов, стоит помнить, что он зависит от общего количества откартированных ридов .
Показатель NRF уменьшается с увеличением глубины секвенирования библиотеки. При это в конечном итоге достигается точка, в которой сложность будет максимальна и будет происходить секвенирование одних и тех же фрагментов ДНК, амплифицированных с помощью ПЦР . Низкая сложность библиотеки может возникнуть, например, если в процессе иммунопреципитации выделяется очень небольшое количество ДНК .
Чувствительность
Чувствительность технологии зависит от глубины секвенирования, длины генома и других факторов. Для транскрипционных факторов млекопитающих и энхансер-ассоциированных модификаций хроматина, которые обычно локализованы в специфических узких сайтах и имеют порядка тысячи сайтов связывания, будет достаточно около 20 миллионов чтений . Для белков с бо́льшим числом сайтов связывания ( РНК-полимераза III ) потребуется до 60 миллионов чтений . В случае транскрипционных факторов червей или мушек необходимо примерно 4 миллиона чтений . Цена секвенирования полученных после иммунопреципитации фрагментов непосредственно коррелирует с глубиной секвенирования. Если требуется отобразить с высокой чувствительностью участки связывания белков, часто встречающиеся в большом геноме, потребуются высокие затраты, так как необходимо будет большое число чтений. Это отличает данный метод от ChIP-on-Chip, в котором чувствительность не связана со стоимостью анализа .
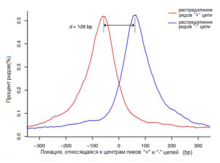
Ещё одно отличие от ChIP-методов, основанных на ДНК-микрочипах, заключается в том, что точность ChIP-seq не ограничивается расстоянием между заданными зондами. Путём интеграции большого количества коротких чтений может быть получена локализация сайтов связывания с высокой точностью. В сравнении с методами ChIP-on-Chip данные ChIP-seq могут быть использованы для локализации фактического сайта связывания белка с точностью до десятков нуклеотидов. Плотность чтений на участках связывания является хорошим индикатором силы связи белок-ДНК, что позволяет легче количественно оценивать и сравнивать сродство белка к разным участкам .
Точность и специфичность
Длина типичного участка связывания белка составляет 6—20 нуклеотидов, а длина полученных фрагментов после ChIP — около 200, что делает определение места связывания не слишком точным. Кроме того, полученные библиотеки часто могут содержать участки ДНК, не связанной с исследуемым белком, что приводит к ошибкам в результатах. Существуют различные модификации метода, направленные на повышение точности (например, ChIP-exo). Качество эксперимента ChIP-seq также прямо зависит от специфичности антител и степени обогащения образца на стадии иммунопреципитации. Главными проблемами могут быть низкая реактивность антитела против нужного белка и/или кросс-реактивность по отношению к другим белкам. Консорциум ENCODE предлагает несколько методик для оценки специфичности антител .
Для осуществления иммунопреципитации также можно пришить к исследуемому белку эпитоп . Такой способ решает обе проблемы, возникающие при иммунопреципитации антителами, однако в этом случае пришиваемый тэг может влиять на исследуемый белок (например, изменять уровень его экспрессии или способность связывания) .
Альтернативные методы
ChIP-on-chip
, сочетающий иммунопреципитацию хроматина с гибридизацией на ДНК-микрочипах , ранее был самым популярным методом для установления ДНК-белковых взаимодействий. Chip-seq и ChIP-on-chip — два наиболее широко распространённых подхода в полногеномных исследованиях взаимодействий ДНК — белок in vivo. Однако при более детальном сравнении этих методов удаётся показать значительные преимущества Chip-seq . Сравнение методов Chip-seq и ChIP-on-Chip представлено в таблице :
| Показатель | ChIP-seq | ChIP-on-Chip |
|---|---|---|
| Количество исходной ДНК | менее 10 нг | 4 мкг |
| Гибкость метода | да: полногеномный анализ любого отсеквенированного организма | есть ограничения: доступность ДНК-микрочипов |
| Точность определения позиции участка связывания | +/- 50 пн | +/- 500 − 1000 пн |
| Чувствительность | вариабельная: увеличивая количество чтений, можно увеличить чувствительность | слабая: зависит от качества гибридизации |
| Кросс-гибридизация (гибридизация одноцепочечной ДНК с зондом, который ей частично комплементарен) | исключена: каждая молекула ДНК секвенируется отдельно | может быть значительной, что сильно снижает точность анализа |
DamID
(DNA adenine methyltransferase identification) позволяет картировать сайты ДНК-белковых взаимодействий в клетках эукариот. Для этого в клетках экспрессируется химерный белок , состоящий из интересующего белка и ДНК аденин метилтрансферазы (Dam) E. coli , которая метилирует аденины в последовательности GATC. У большинства эукариот эндогенное метилирование аденина в сайтах GATC не происходит. Когда исследуемый белок, слитый с Dam, связывается с ДНК или другими ДНК-ассоциированными белками, Dam метилирует остатки аденина в ДНК, окружающей сайт связывания, таким образом данный метод позволяет маркировать сайты взаимодействия целевого белка с ДНК и ДНК-ассоциированными белками. Чтобы идентифицировать последовательности, метилированные химерным белком, метилированные фрагменты селективно амплифицируются и гибридизуются на микрочипах .
Селективная амплификация метилированных фрагментов ДНК основана на специальном ПЦР-протоколе. Сначала ДНК, метилированная в сайтах GATC, разрезается между GA m and TC нуклеотидами рестриктазой . Расщепление с помощью DpnI приводит к образованию фрагментов ДНК с тупыми концами 5’ TC и 3’ GA m . После этого к полученным фрагментам лигируются двухцепочечные адапторы. Продукты лигирования затем расщепляются эндонуклеазой рестрикции . DpnII разрезает ДНК по неметилированным сайтам GATC, благодаря этому впоследствии амплифицируются только фрагменты, фланкированные последовательно метилированными сайтами GATC (то есть сайтами, между которыми не встречаются неметилированные сайты GATC). Затем проводится ПЦР с праймерами, комплементарными к адапторам, и таким образом специфично амплифицируются геномные фрагменты с метилированными сайтами GATC по краям .
Модификации метода
Со времени изобретения ChIP-Seq было придумано множество модификаций этого метода, которые позволяют более эффективно выполнять те или иные подзадачи.
ChIA-PET
Данный метод применяется для определения взаимодействий участков хроматина, расположенных на значительном удалении друг от друга в геноме . В основе ChIA-PET лежит теория проксимального лигирования ( англ. proximity ligation), гласящая, что концы участков хроматина, связанных с белковым комплексом, находящиеся рядом, будут лигироваться друг на друга с большей вероятностью, чем концы участков, находящихся в растворе или связанных с другим белковым комплексом.
PLAC-seq
Существует множество методов исследования дальних взаимодействий хроматина, но они требуют большого количества клеток для анализа. Для преодоления этого ограничения был разработан метод PLAC-seq (Proximity Ligation-Assisted ChIP-seq), в котором сшивка сближенных участков осуществляется в ядре до фрагментации хроматина и иммунопреципитации. PLAC-seq демонстрирует лучшую точность, эффективность и воспроизводимость по сравнению с ChIA-PET при определении дальних контактов в клетках млекопитающих .
Nano-ChIP-seq
Метод nano-ChIP-seq основан на том, что выделенная в ходе эксперимента ChIP ДНК амплифицируется с помощью ПЦР и уже после этого секвенируется . Это позволяет проводить анализ на малом количестве клеток, обычно около 10 тысяч. Тем не менее, достаточное число клеток зависит от множества факторов, таких как эффективность антител и обогащённость образца целевым белком, поэтому в каких-то случаях может понадобиться больше 10 тысяч клеток .
ChIP-exo и ChIP-nexus
Метод — модификация протокола ChIP-seq, позволяющая улучшить разрешение найденных сайтов связывания от сотен пар оснований практически до одного нуклеотида. В ChIP-exo используется для удаления контаминирующей ДНК и 5′-концов сшитых с целевым белком фрагментов ДНК вплоть до позиции на каком-то фиксированном расстоянии от места связывания белка . Так как в результате эксперимента ChIP образуются ДНК-фрагменты обеих цепей, выровненные 5′ концы картируются на две позиции генома, между которыми и находится сайт связывания белка. Эксперименты на дрожжах показали, что ChIP-exo позволяет идентифицировать сайты связывания с нуклеотидной точностью и в 40 раз бо́льшим отношением сигнал-шум по сравнению с ChIP-seq и ChIP-on-Chip .
Модификацией протокола ChIP-exo является протокол ChIP-nexus (ChIP experiments with nucleotide resolution through exonuclease, unique barcode and single ligation). В этом протоколе к ДНК лигируются специальные адапторы, которые содержат пару последовательностей для амплификации библиотеки, сайт рестриктазы BamHI и рандомизированный баркод , который позволяет отследить избыточную амплификацию фрагментов. Так же, как и в протоколе ChIP-exo, проводится обработка λ-экзонуклеазой, которая расщепляет ДНК с 5'-конца до физического препятствия в виде связанного с ДНК белка. После этого проводится внутримолекулярная циркуляризация ДНК, а затем релинеаризация путём обработки рестриктазой BamHI . Таким образом по краям интересующего фрагмента оказываются последовательности для амплификации. Этот дополнительный шаг позволяет улучшить эффективность встраивания ДНК-фрагментов в библиотеку .
Competition-ChIP
Competition-ChIP — модификация протокола ChIP-seq, использующаяся для измерения относительной динамики связывания транскрипционных факторов с ДНК . Идея метода основана на экспрессии двух копий изучаемого транскрипционного фактора с разными эпитопными метками . Одна из этих копий экспрессируется на постоянной основе, а экспрессия второй, выступающей в качестве конкурента, индуцибельна. Соотношение изоформ, ассоциированных с определёнными локусами, определяется либо с помощью ChIP-seq, либо ChIP-on-chip. Скорость замены конститутивно экспрессирующейся формы на индуцибельную позволяет рассчитать время пребывания исследуемого фактора на каждом сайте связывания.
CLIP-seq
(также известный как HITS-CLIP — high-throughput sequencing of RNA isolated by crosslinking immunoprecipitation) — метод исследования РНК-белковых взаимодействий и модификаций РНК in vivo .
DRIP-seq и DRIVE-seq
R-петли — трехцепочечные структуры, образованные вытесненной одноцепочечной ДНК (оцДНК) и дуплексом РНК-оцДНК. In vivo на их долю приходится примерно 5-8 % генома. Через регуляцию связывания различных белков R-петли участвуют во многих клеточных процессах, таких как, например, дифференциация эмбриональных стволовых клеток . Для исследования R-петель был разработан метод (DNA:RNA ImmunoPrecipitation and sequencing), который по сути очень похож на ChIP-Seq, но основан на использовании специфичных к R-петлям антител . Другим способом исследования R-петель является метод DRIVE-seq (DNA:RNA In Vitro Enrichment and sequencing), в котором вместо антител используется инактивированная эндонуклеаза MBP-RNASEH1 . DRIVE-seq может быть использован для уточнения предсказаний, полученных с помощью DRIP-seq. Оба метода позволяют точно и практически количественно оценить число R-петель. Впервые DRIP-seq был использован для изучения R-петель в геноме человека: было показано, что большое их количество содержится в промоторов .
CETCh-seq
Метод CETCh-seq создан для того, чтобы при изучении ДНК-белковых взаимодействий преодолеть такую техническую проблему, как доступность подходящих для ChIP-seq экспериментов антител. С помощью геномного редактирования с использованием CRISPR/Cas9 к интересующим белкам, например, транскрипционным факторам, присоединяется эпитоп для дальнейшего распознавания подходящими антителами .
CUT&RUN
— модификация ChIP-seq, позволяющая сильно поднять соотношение сигнал/шум. Эффект достигается за счёт использования микрококковой нуклеазы , слитой с белком А , на стадии иммунопреципитации .
CUT&Tag
— метод, аналогичный CUT&RUN, однако вместо микрококковой нуклеазы используется транспозаза Tn5. Преимущество этого метода над CUT&RUN заключается в том, что он не требует лизирования клеток и фракционирования хроматина .
Применение
ChIP-seq в принципе применим для любых белков, которые осаждаются в ходе иммунопреципитации хроматина. Типичным примером использования метода ChIP-seq является определение участков связывания транскрипционных факторов, ДНК-полимеразы , структурных белков, а также модификаций гистонов и структуры хроматина . В качестве альтернативы ChIP-seq был разработан ряд не использующих иммунопреципитацию методов ( DNase-Seq и FAIRE-Seq ) для определения свободных от нуклеосом участков ДНК .
Поиск мотивов
Одной из основных целей ChIP-seq экспериментов является поиск в последовательности ДНК мотивов для связывания белков. Участки ДНК, физически контактирующие с факторами транскрипции и другими белками, могут быть изолированы методом иммунопреципитации хроматина. В ходе эксперимента получается набор фрагментов ДНК, связанных с исследуемым белком in vivo . Дальнейший анализ включает использование массивного параллельного секвенирования и баз данных полных геномов для определения положения участков связывания в геноме . Наиболее широко используемый инструмент для обнаружения мотивов — алгоритм MEME (Multiple EM for Motif Elicitation). Часто множество мотивов может быть найдено на основе одного датасета и анализ мотивов может быть проведён даже на ChIP-seq данных низкого качества, но значимость и достоверность таких мотивов будет ниже .
Поиск участков с биологической функцией
Данные экспериментов ChIP-seq часто используются для определения регуляторных участков для интересующего локуса . В частности, ChIP-seq широко используется для изучения бактериальных регулонов . Для этого после нахождения участков связывания производится поиск предполагаемых регулируемых генов .
Дифференциальный анализ
Определение различий между профилями ChIP-Seq при разных условиях производится после вызова пиков. Пики, полученные в разных экспериментах, сливаются затем в один список. Для дальнейшего определения участков-кандидатов часто используются программы для анализа дифференциальной экспрессии генов , например, DESeq2 и edgeR . Эти программы способны проводить дифференциальный анализ, обрабатывая списки полученных пиков как списки «генов». Существуют также программы, разработанные специально для дифференциального анализа данных ChIP-Seq (например, DiffBind , ChIPComp , DBChIP ), которые работают по схожему принципу. Многие другие программы (например, PePr ) используют модели, не требующие предварительного вызова пиков .
Исследование состояния хроматина
Метилирование ДНК и модификации гистонов претерпевают сильные изменения в процессе переходов между стадиями развития и при заболеваниях, таких как рак, и таким образом вносят основной вклад в динамическую природу хроматина. Различные модификации гистонов исследуются с использованием специфичных антител, чтобы получить профиль гистоновых меток в образце. В собственных экспериментах консорциум ENCODE тщательно тестирует специфичность используемых антител на множестве различно модифицированных гистоновых концевых пептидов. Используются также общие источники клеток, которые профилированы и сравнены, для обеспечения согласованности между экспериментами. Современные методические рекомендации консорциума ENCODE охватывают валидацию антител, воспроизводимость экспериментов, глубину секвенирования, анализ качества данных, публикацию данных и метаданных .
Анализ аллельного дисбаланса
Всё больший интерес вызывает анализ данных ChIP-Seq со внутренним контролем по другому аллелю для выявления . При этом данные, полученные из эксперимента ChIP-Seq, используются для поиска связи биологических сигналов с однонуклеотидными полиморфизмами (SNP) . Данный анализ включает три стадии :
- выравнивание ридов, то есть определение позиции в геноме и аллеля для каждого рида,
- подсчёт числа достоверно откартированных ридов для каждого SNP для каждого аллеля,
- ранжирование возможных SNP и статистическая оценка аллельного дисбаланса.
Для первых двух этапов важна правильная стратегия картирования ридов на референсный геном, так как необходимо отличать ошибки секвенирования от реально существующих аллелей. Для третьего этапа разработано несколько программ, использующих разные статистические тесты, например, AlleleDB , NPBin и WASP .

Базы данных
Геном многоклеточных организмов крайне сложен, и не до конца понятно в деталях, как происходит реализация наследственной информации. Детальное понимание работы генома требует наличие полного списка функциональных элементов и описания того, как они действуют в течение времени и в различных типах клеток. В попытке решения данной проблемы были созданы проекты ENCODE и modENCODE . Помимо результатов ChIP-seq, в ENCODE и modENCODE интегрируются данные таких анализов, как 5С и ChIA-PET , позволяющих определить конформацию хромосом; DNase-seq и FAIRE-Seq , позволяющих определить свободные от нуклеосом участки; бисульфитного секвенирования и , позволяющих определить наличие метилцитозинов в ДНК, RT-PCR и секвенирования РНК , позволяющих определить уровень экспрессии генов, а также и , позволяющих выявить РНК -белковые взаимодействия .
По состоянию на второе десятилетие XXI века существует ряд баз данных, содержащих результаты экспериментов ChIP-seq и их анализа:
- ENCODE — на проекта можно скачать координаты участков связывания ДНК с транскрипционными факторами или модифицированными гистонами, полученными в результате ChIP-seq. Содержит данные по различным клеточным линиям и тканям мыши и человека .
- — база данных для визуализации и использования общедоступных данных ChIP-seq. ChIP-Atlas охватывает почти все общедоступные данные ChIP-seq, представленные в SRA (Sequence Read Archives) в NCBI, DDBJ или ENA, и основан на более чем 118 000 экспериментов.
- — проект, посвящённый анализу ДНК-элементов плодовой мушки D. melanogaster и нематоды C. elegans .
- — база данных, сгенерированная на основе ENCODE .
- — помимо человека и мыши, доступны результаты экспериментов ChIP-seq собаки, курицы, дрозофилы и нематоды C. elegans .
- — ChIP-seq человека, мыши и крысы, можно получить список участков связывания с различными белками, в которые попал исследуемый ген .
- — база данных участков связывания инсулятора CTCF .
- — ChIP-seq и ChIP-chip человека и мыши .
- — база данных участков связывания транскрипционных факторов человека .
- — профили участков связывания транскрипционных факторов на основе ChIP-seq различных эукариот .
- — база данных аннотированных регуляторных сайтов .
- — ChIP-Seq и DNase-Seq человека и мыши .
- — интегрированная база данных регуляторов хроматина, доступны результаты экспериментов ChIP-seq человека и мыши .
Исследования
Эукариоты
В качестве примера успешного использования ChIP-seq для изучения эукариот можно привести исследование нуклеосомной архитектуры промоторов . С помощью ChIP-seq удалось установить, что у дрожжей, возможно, имеются свободные от нуклеосом области промоторов (длиной примерно 150 н. п.), с которых РНК-полимераза может инициировать транскрипцию . Данный метод также был успешно применён для поиска сайтов связывания 22 транскрипционных факторов в геноме нематоды C. elegans . Для 20 % всех аннотированных генов генома нематоды были определены регулирующие их факторы транскрипции .
ChIP-seq также широко используется для изучения модификаций гистонов. Известно более 100 модификаций гистонов . Наример, известно, что ацетилирование, в частности, ацетилирование лизина 9 гистона Н3 (H3K9Ac), обычно ассоциируется с открытыми и доступными областями хроматина ( эухроматином ). В то же время метилирование гистонов может быть ассоциировано как с открытыми, так и с плотно упакованными областями хроматина ( гетерохроматином ). В частности, моно- и триметилирование лизина 4 гистона Н3 (H3K4me1 или H3K4me3) обычно ассоциируется с открытым хроматином, причём каждая из этих меток представляет особую категорию открытого хроматина: H3K4me3 маркирует промоторные регионы, H3K4me1 — маркирует транскрипционные энхансеры, H3K36me3 — маркирует транскрибируемые участки генома. Триметилирование лизинов 9 и 27 гистона Н3 (H3K9me3 и H3K27me3), напротив, ассоциируется с компактизацией хроматина и, как следствие, репрессией генов. H3K9me3 и H3K27me3 регулируют разные типы генов: H3K27me3 преимущественно подавляет гомеобоксные транскрипционные факторы, а целевыми генами H3K9me3 преимущественно являются транскрипционные факторы с мотивом «цинковые пальцы» () . Различные комбинации гистоновых меток могут обеспечить ещё более подробную информацию: например, присутствие сразу двух меток H3K4me3 (метки эухроматина) и H3K9me3 (метки гетерохроматина) на промоторе может быть идентификатором импринтируемых генов .
Прокариоты
У бактерий регуляция экспрессии генов на уровне транскрипции осуществляется с помощью транскрипционных факторов . Метод ChIP-seq может быть использован для определения участков связывания таких транскрипционных факторов. Некоторые бактериальные транскрипционные факторы имеют несколько сайтов связывания внутри промотора (то есть сайтов, расположенных на расстоянии менее 100 п.н.) . Большинство алгоритмов поиска пиков определяют такие близко расположенные сайты как один. Для решения этой проблемы используются так называемые алгоритмы деконволюции пиков, например, CSDeconv , GEM , PICS или dPeak .
Следующим шагом после определения сайтов связывания является определение регулируемых генов. Обычно ассоциация найденных пиков с генами выполняется алгоритмически с помощью поиска близлежащих сайтов старта транскрипции (transcription start site, TSS). Однако в случае бактерий (в том числе E. coli ) TSS могут быть не определены для многих генов, поэтому вместо TSS можно искать близлежащие сайты старта трансляции, вручную исследовать геномное окружение пика или использовать данные экспрессии генов (например, сравнивать экспрессию регулонов в диком типе и при делеции исследуемого транскрипционного фактора на основании данных RNA-seq) .
Перспективы развития
Текущие успехи метода ChIP-seq уже позволяют анализировать образцы, содержащие гораздо меньше клеток, что значительно расширяет его применимость в таких областях, как эмбриология и биология развития, где получать большие образцы слишком дорого или трудно. Метод определённо имеет потенциал для обнаружения мутаций в сайтах связывания, которые влияют на связывание с белками и регуляцию экспрессии генов .
Однако становится очевидным, что проблемы ChIP-seq требуют новых экспериментальных, статистических и вычислительных решений. Необходимо снизить количество артефактов и ложно-положительных результатов, а также научиться отличать индивидуальные эффекты изучаемых явлений от контекстно-зависимых. Важные новые разработки связаны с обнаружением и анализом дистальных (находящихся на значительном расстоянии от гена) регуляторных областей. Возможно, с помощью ChIP-seq можно будет определять непрямое связывание ДНК, например, через дополнительные белки или комплексы белков, так как предсказанные сайты могут быть функциональными вне зависимости от наличия специфического мотива. Наконец, необходимо использовать дополнительную информацию (например, уровень экспрессии или данные о конформации хроматина), чтобы отличать реальную функциональность, так как связывание с ДНК не обязательно подразумевает определённую функцию .
Перспективным направлением является интеграция данных, полученных из большого числа экспериментов, для разрешения и анализа сложных взаимодействий. Для этой цели часто применяются различные методы машинного обучения .
Примечания
- Mikkelsen T. S. , Ku M. , Jaffe D. B. , Issac B. , Lieberman E. , Giannoukos G. , Alvarez P. , Brockman W. , Kim T. K. , Koche R. P. , Lee W. , Mendenhall E. , O'Donovan A. , Presser A. , Russ C. , Xie X. , Meissner A. , Wernig M. , Jaenisch R. , Nusbaum C. , Lander E. S. , Bernstein B. E. (англ.) // Nature. — 2007. — Vol. 448, no. 7153 . — P. 553—560. — doi : . — .
- Barski A. , Cuddapah S. , Cui K. , Roh T. Y. , Schones D. E. , Wang Z. , Wei G. , Chepelev I. , Zhao K. (англ.) // Cell. — 2007. — Vol. 129, no. 4 . — P. 823—837. — doi : . — .
- Johnson D. S. , Mortazavi A. , Myers R. M. , Wold B. (англ.) // Science (New York, N.Y.). — 2007. — Vol. 316, no. 5830 . — P. 1497—1502. — doi : . — .
- ↑ Park P. J. (англ.) // Nature reviews. Genetics. — 2009. — Vol. 10, no. 10 . — P. 669—680. — doi : . — .
- ↑ Barbara Kaboord, Maria Perr. (англ.) // Methods in Molecular Biology (Clifton, N.J.). — 2008-01-01. — Vol. 424 . — P. 349–364 . — ISSN . — doi : . 23 апреля 2017 года.
- ↑ Terrence S. Furey. (англ.) // Nature Reviews. Genetics. — 2012-12-01. — Vol. 13 , iss. 12 . — P. 840–852 . — ISSN . — doi : . 23 апреля 2017 года.
- ↑ Ryuichiro Nakato, Katsuhiko Shirahige. (англ.) // Briefings in Bioinformatics. — 2016-03-15. — P. bbw023 . — ISSN . — doi : . 21 января 2022 года.
- ↑ Timothy Bailey, Pawel Krajewski, Istvan Ladunga, Celine Lefebvre, Qunhua Li. (англ.) // PLoS computational biology. — 2013-01-01. — Vol. 9 , iss. 11 . — P. e1003326 . — ISSN . — doi : . 4 мая 2017 года.
- Anthony M. Bolger, Marc Lohse, Bjoern Usadel. (англ.) // Bioinformatics. — 2014-08-01. — Vol. 30 , iss. 15 . — P. 2114–2120 . — ISSN . — doi : . 24 апреля 2017 года.
- ↑ Joel Rozowsky, Ghia Euskirchen, Raymond K Auerbach, Zhengdong D Zhang, Theodore Gibson. (англ.) // Nature Biotechnology. — 2009-1. — Vol. 27 , iss. 1 . — P. 66–75 . — ISSN . — doi : . 30 марта 2019 года.
- Heng Li, Bob Handsaker, Alec Wysoker, Tim Fennell, Jue Ruan. (англ.) // Bioinformatics. — 2009-08-15. — Vol. 25 , iss. 16 . — P. 2078–2079 . — ISSN . — doi : . 24 апреля 2017 года.
- Hashem Koohy, Thomas A. Down, Mikhail Spivakov, Tim Hubbard. // PLoS ONE. — 2014-05-08. — Т. 9 , вып. 5 . — С. e96303 . — ISSN . — doi : .
- Elizabeth G. Wilbanks, Marc T. Facciotti. // PLoS ONE. — 2010-07-08. — Т. 5 , вып. 7 . — С. e11471 . — ISSN . — doi : .
- Teemu D Laajala, Sunil Raghav, Soile Tuomela, Riitta Lahesmaa, Tero Aittokallio. // BMC Genomics. — 2009. — Т. 10 , вып. 1 . — С. 618 . — ISSN . — doi : .
- ↑ S. G. Landt, G. K. Marinov, A. Kundaje, P. Kheradpour, F. Pauli. (англ.) // Genome Research. — 2012-09-01. — Vol. 22 , iss. 9 . — P. 1813–1831 . — ISSN . — doi : .
- Assaf Rotem, Oren Ram, Noam Shoresh, Ralph A. Sperling, Alon Goren. // Nature biotechnology. — 2015-11. — Т. 33 , вып. 11 . — С. 1165–1172 . — ISSN . — doi : . 21 мая 2016 года.
- The ENCODE Project Consortium. (англ.) // PLoS Biology / Peter B. Becker. — 2011-04-19. — Vol. 9 , iss. 4 . — P. e1001046 . — ISSN . — doi : .
- ↑ Joshua W. K. Ho, Eric Bishop, Peter V. Karchenko, Nicolas Nègre, Kevin P. White. (англ.) // BMC genomics. — 2011-02-28. — Vol. 12 . — P. 134 . — ISSN . — doi : . 4 мая 2017 года.
- Frauke Greil, Celine Moorman, Bas van Steensel. DamID: Mapping of In Vivo Protein–Genome Interactions Using Tethered DNA Adenine Methyltransferase] (англ.) // Methods in Enzymology. — Elsevier, 2006. — Vol. 410 . — P. 342–359 . — ISBN 9780121828158 . — doi : . 12 мая 2019 года.
- Bas van Steensel, Daniel Peric-Hupkes, Maartje J. Vogel. (англ.) // Nature Protocols. — 2007-06. — Vol. 2 , iss. 6 . — P. 1467–1478 . — ISSN . — doi : . 25 мая 2021 года.
- Yi Eve Sun, Weihong Ge. (неопр.) F1000 - Post-publication peer review of the biomedical literature (4 декабря 2009). Дата обращения: 18 апреля 2020.
- Rongxin Fang, Miao Yu, Guoqiang Li, Sora Chee, Tristin Liu. (англ.) // Cell Research. — 2016-12. — Vol. 26 , iss. 12 . — P. 1345–1348 . — ISSN . — doi : . 30 марта 2019 года.
- ↑ Mazhar Adli, Bradley E Bernstein. (англ.) // Nature Protocols. — 2011-10. — Vol. 6 , iss. 10 . — P. 1656–1668 . — ISSN . — doi : . 18 апреля 2019 года.
- ↑ Ho Sung Rhee, B. Franklin Pugh. (англ.) // Cell. — 2011-12. — Vol. 147 , iss. 6 . — P. 1408–1419 . — doi : . 18 апреля 2019 года.
- ↑ Qiye He, Jeff Johnston, Julia Zeitlinger. // Nature biotechnology. — 2015-4. — Т. 33 , вып. 4 . — С. 395–401 . — ISSN . — doi : .
- Colin R Lickwar, Florian Mueller, Jason D Lieb. (англ.) // Nature Protocols. — 2013-7. — Vol. 8 , iss. 7 . — P. 1337–1353 . — ISSN . — doi : . 20 апреля 2019 года.
- Robert B. Darnell. (англ.) // Wiley Interdisciplinary Reviews: RNA. — 2010-9. — Vol. 1 , iss. 2 . — P. 266–286 . — ISSN . — doi : . 20 апреля 2019 года.
- László Halász, Zsolt Karányi, Beáta Boros-Oláh, Tímea Kuik-Rózsa, Éva Sipos. (англ.) // Genome Research. — 2017-6. — Vol. 27 , iss. 6 . — P. 1063–1073 . — ISSN . — doi : .
- ↑ Paul A. Ginno, Paul L. Lott, Holly C. Christensen, Ian Korf, Frédéric Chédin. (англ.) // Molecular Cell. — 2012-3. — Vol. 45 , iss. 6 . — P. 814–825 . — doi : . 20 апреля 2019 года.
- Daniel Savic, E. Christopher Partridge, Kimberly M. Newberry, Sophia B. Smith, Sarah K. Meadows. (англ.) // Genome Research. — 2015-10. — Vol. 25 , iss. 10 . — P. 1581–1589 . — ISSN . — doi : .
- Peter J Skene, Steven Henikoff. (англ.) // eLife. — 2017-01-16. — Vol. 6 . — P. e21856 . — ISSN . — doi : . 13 мая 2020 года.
- M. Robyn Andersen, Kelsey Afdem, Marcia Gaul, Shelly Hager, Erin Sweet. // OBM Genetics. — 2019-02-27. — Т. 3 , вып. 3 . — С. 1–1 . — ISSN . — doi : .
- ↑ (неопр.) . epigenie.com. Дата обращения: 22 апреля 2019. 22 апреля 2019 года.
- ↑ Kevin S. Myers, Dan M. Park, Nicole A. Beauchene, Patricia J. Kiley. (англ.) // Methods. — 2015-9. — Vol. 86 . — P. 80–88 . — doi : . 2 мая 2019 года.
- Michael I Love, Wolfgang Huber, Simon Anders. // Genome Biology. — 2014-12. — Т. 15 , вып. 12 . — ISSN . — doi : .
- M. D. Robinson, D. J. McCarthy, G. K. Smyth. // Bioinformatics. — 2009-11-11. — Т. 26 , вып. 1 . — С. 139–140 . — ISSN . — doi : .
- Anaïs Bardet. // Practical Guide to ChIP-seq Data Analysis. — CRC Press, 2018-10-26. — С. 41–52 . — ISBN 9780429487590 .
- Li Chen, Chi Wang, Zhaohui S. Qin, Hao Wu. // Bioinformatics. — 2015-02-13. — Т. 31 , вып. 12 . — С. 1889–1896 . — ISSN . — doi : .
- Kun Liang, Sündüz Keleş. // Bioinformatics. — 2011-11-03. — Т. 28 , вып. 1 . — С. 121–122 . — ISSN . — doi : .
- ↑ Yanxiao Zhang, Yu-Hsuan Lin, Timothy D. Johnson, Laura S. Rozek, Maureen A. Sartor. // Bioinformatics. — 2014-06-03. — Т. 30 , вып. 18 . — С. 2568–2575 . — ISSN . — doi : .
- Bradley E Bernstein, John A Stamatoyannopoulos, Joseph F Costello, Bing Ren, Aleksandar Milosavljevic. // Nature biotechnology. — 2010-10. — Т. 28 , вып. 10 . — С. 1045–1048 . — ISSN . — doi : . 22 мая 2016 года.
- ↑ Qi Zhang, Sündüz Keleş. // Biostatistics. — 2017-11-03. — Т. 19 , вып. 4 . — С. 546–561 . — ISSN . — doi : .
- Qi Zhang. (англ.) // Computational Epigenetics and Diseases. — Elsevier, 2019. — P. 67–77 . — ISBN 9780128145135 . — doi : . 5 мая 2019 года.
- Christopher Gregg. (неопр.) F1000 - Post-publication peer review of the biomedical literature (11 июля 2016). Дата обращения: 5 мая 2019.
- Bryce van de Geijn, Graham McVicker, Yoav Gilad, Jonathan K Pritchard. // Nature Methods. — 2015-09-14. — Т. 12 , вып. 11 . — С. 1061–1063 . — ISSN . — doi : .
- ↑ Susan E. Celniker, Laura A. L. Dillon, Mark B. Gerstein, Kristin C. Gunsalus, Steven Henikoff. (англ.) // Nature. — 2009-06-18. — Vol. 459 , iss. 7249 . — P. 927–930 . — ISSN . — doi : . 29 апреля 2017 года.
- Hongzhu Qu, Xiangdong Fang. (англ.) // Genomics, Proteomics & Bioinformatics. — 2013-06-01. — Vol. 11 , iss. 3 . — P. 135–141 . — ISSN . — doi : . 5 мая 2017 года.
- Oki, S; Ohta, T. . — 2015. — doi : .
- modENCODE Consortium, Sushmita Roy, Jason Ernst, Peter V. Kharchenko, Pouya Kheradpour. (англ.) // Science (New York, N.Y.). — 2010-12-24. — Vol. 330 , iss. 6012 . — P. 1787–1797 . — ISSN . — doi : . 5 мая 2017 года.
- Jie Wang, Jiali Zhuang, Sowmya Iyer, Xin-Ying Lin, Melissa C. Greven. (англ.) // Nucleic Acids Research. — 2013-01-01. — Vol. 41 , iss. Database issue . — P. D171–176 . — ISSN . — doi : . 5 мая 2017 года.
- Jian-Hua Yang, Jun-Hao Li, Shan Jiang, Hui Zhou, Liang-Hu Qu. (англ.) // Nucleic Acids Research. — 2013-01-01. — Vol. 41 , iss. Database issue . — P. D177–187 . — ISSN . — doi : . 5 мая 2017 года.
- Alexander Lachmann, Huilei Xu, Jayanth Krishnan, Seth I. Berger, Amin R. Mazloom. (англ.) // Bioinformatics (Oxford, England). — 2010-10-01. — Vol. 26 , iss. 19 . — P. 2438–2444 . — ISSN . — doi : . 5 мая 2017 года.
- Jesse D. Ziebarth, Anindya Bhattacharya, Yan Cui. (англ.) // Nucleic Acids Research. — 2013-01-01. — Vol. 41 , iss. Database issue . — P. D188–194 . — ISSN . — doi : . 5 мая 2017 года.
- Li Chen, George Wu, Hongkai Ji. (англ.) // Bioinformatics (Oxford, England). — 2011-05-15. — Vol. 27 , iss. 10 . — P. 1447–1448 . — ISSN . — doi : . 5 мая 2017 года.
- Ivan V. Kulakovskiy, Ilya E. Vorontsov, Ivan S. Yevshin, Anastasiia V. Soboleva, Artem S. Kasianov. (англ.) // Nucleic Acids Research. — 2016-01-04. — Vol. 44 , iss. D1 . — P. D116–125 . — ISSN . — doi : . 5 мая 2017 года.
- Albin Sandelin, Wynand Alkema, Pär Engström, Wyeth W. Wasserman, Boris Lenhard. (англ.) // Nucleic Acids Research. — 2004-01-01. — Vol. 32 , iss. Database issue . — P. D91–94 . — ISSN . — doi : . 5 мая 2017 года.
- Mikhail Pachkov, Piotr J. Balwierz, Phil Arnold, Evgeniy Ozonov, Erik van Nimwegen. (англ.) // Nucleic Acids Research. — 2013-01-01. — Vol. 41 , iss. Database issue . — P. D214–220 . — ISSN . — doi : . 5 мая 2017 года.
- Bo Qin, Meng Zhou, Ying Ge, Len Taing, Tao Liu. (англ.) // Bioinformatics (Oxford, England). — 2012-05-15. — Vol. 28 , iss. 10 . — P. 1411–1412 . — ISSN . — doi : . 5 мая 2017 года.
- Qixuan Wang, Jinyan Huang, Hanfei Sun, Jing Liu, Juan Wang. (англ.) // Nucleic Acids Research. — 2014-01-01. — Vol. 42 , iss. Database issue . — P. D450–458 . — ISSN . — doi : . 5 мая 2017 года.
- Christoph D. Schmid, Philipp Bucher. (англ.) // Cell. — 2007-11-30. — Vol. 131 , iss. 5 . — P. 831–832; author reply 832–833 . — ISSN . — doi : . 5 мая 2017 года.
- Wei Niu, Zhi John Lu, Mei Zhong, Mihail Sarov, John I. Murray. (англ.) // Genome Research. — 2011-02-01. — Vol. 21 , iss. 2 . — P. 245–254 . — ISSN . — doi : . 5 мая 2017 года.
- Xiong Ji, Daniel B. Dadon, Brian J. Abraham, Tong Ihn Lee, Rudolf Jaenisch. // Proceedings of the National Academy of Sciences. — 2015-03-09. — С. 201502971 . — ISSN . — doi : .
- Huihuang Yan, Shulan Tian, Susan L Slager, Zhifu Sun. (англ.) // Epigenomics. — 2016-9. — Vol. 8 , iss. 9 . — P. 1239–1258 . — ISSN . — doi : .
- Henriette O’Geen, Lorigail Echipare, Peggy J. Farnham. // Methods in molecular biology (Clifton, N.J.). — 2011. — Т. 791 . — С. 265–286 . — ISSN . — doi : .
- Tarjei S. Mikkelsen, Manching Ku, David B. Jaffe, Biju Issac, Erez Lieberman. // Nature. — 2007-08-02. — Т. 448 , вып. 7153 . — С. 553–560 . — ISSN . — doi : . 22 мая 2016 года.
- Douglas F. Browning, Stephen J. W. Busby. // Nature Reviews Microbiology. — 2004-01. — Т. 2 , вып. 1 . — С. 57–65 . — ISSN . — doi : .
- Dongjun Chung, Dan Park, Kevin Myers, Jeffrey Grass, Patricia Kiley. // PLoS Computational Biology. — 2013-10-17. — Т. 9 , вып. 10 . — С. e1003246 . — ISSN . — doi : .
- Antonio L.C. Gomes, Thomas Abeel, Matthew Peterson, Elham Azizi, Anna Lyubetskaya. (англ.) // Genome Research. — 2014-10. — Vol. 24 , iss. 10 . — P. 1686–1697 . — ISSN . — doi : .
- Yuchun Guo, Shaun Mahony, David K. Gifford. (англ.) // PLoS Computational Biology / Stein Aerts. — 2012-08-09. — Vol. 8 , iss. 8 . — P. e1002638 . — ISSN . — doi : .
- Xuekui Zhang, Gordon Robertson, Martin Krzywinski, Kaida Ning, Arnaud Droit. (англ.) // Biometrics. — 2011-3. — Vol. 67 , iss. 1 . — P. 151–163 . — doi : .
- Dongjun Chung, Dan Park, Kevin Myers, Jeffrey Grass, Patricia Kiley. (англ.) // PLoS Computational Biology / Roderic Guigo. — 2013-10-17. — Vol. 9 , iss. 10 . — P. e1003246 . — ISSN . — doi : .
- Jason Ernst, Manolis Kellis. // Nature Biotechnology. — 2010-07-25. — Т. 28 , вып. 8 . — С. 817–825 . — ISSN . — doi : .
- Jason Ernst, Pouya Kheradpour, Tarjei S. Mikkelsen, Noam Shoresh, Lucas D. Ward. // Nature. — 2011-03-23. — Т. 473 , вып. 7345 . — С. 43–49 . — ISSN . — doi : .
- Shirley Pepke, Barbara Wold, Ali Mortazavi. // Nature Methods. — 2009-11. — Т. 6 , вып. 11 . — С. S22–S32 . — ISSN . — doi : .
- 2020-12-14
- 1