Ферменты
- 1 year ago
- 0
- 0


Ферме́нты (от лат. «закваска»), или энзи́мы (от греч. , ἔνζυμον « закваска »), — обычно сложные белковые соединения , РНК ( рибозимы ) или их комплексы, ускоряющие химические реакции в живых системах. Каждый фермент, свернутый в определённую структуру, ускоряет соответствующую химическую реакцию : реагенты в такой реакции называются субстратами , а получающиеся вещества — продуктами. Ферменты специфичны к субстратам: АТФ-аза катализирует расщепление только АТФ, а киназа фосфорилазы фосфорилирует только фосфорилазу .
Ферментативная активность может регулироваться активаторами (повышаться) и ингибиторами (понижаться).
Белковые ферменты синтезируются на рибосомах , а РНК — в ядре .
Термины фермент и энзим давно используют как синонимы : первый в основном в русской и немецкой научной литературе, второй — в англо- и франкоязычной.
Наука о ферментах называется энзимологией , а не ферментологией (чтобы не смешивать корни слов латинского и греческого языков).
В конце XVIII — нач. XIX вв. уже было известно, что мясо переваривается желудочным соком , а крахмал превращается в сахар под действием слюны . Однако механизм этих явлений был неизвестен .
В XIX в. Луи Пастер , изучая превращение углеводов в этиловый спирт под действием дрожжей , пришёл к выводу, что этот процесс ( брожение ) катализируется некой жизненной силой ( ферментом ), находящейся в дрожжевых клетках, причём он считал, что эти «силы» неотделимы от структуры живой клетки дрожжей. Эта точка зрения господствовала в науке в течение длительного времени и шла вразрез с господствовавшей тогда теорией брожения Ю. Либиха , согласно которой все процессы брожения представлялись чисто химическими явлениями каталитического характера (будто бы спиртовое брожение происходит вследствие того, что молекулярные колебания разлагающихся частиц дрожжей передаются сахару и сахар начинает распадаться на спирт и углекислый газ; таким образом дрожжи вызывают брожение не при жизни, а только после своей смерти) .
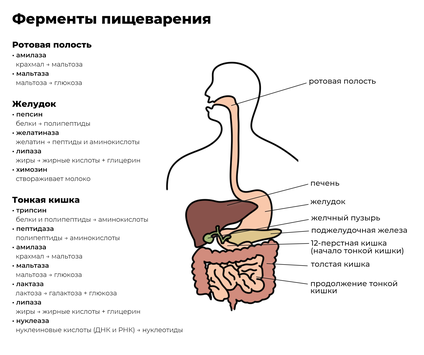
Различные точки зрения о природе спиртового брожения в теоретическом споре Л. Пастера с одной стороны, и механицистов М. Бертло и Ю. Либиха — с другой привели к разделению в научной среде двух соответствующих терминов. Собственно ферментами (от лат. fermentum — закваска) стали называть «организованные ферменты», то есть сами живые микроорганизмы. В противовес этому подходу в 1876 году В. Кюне предложил термин энзим (от греч. ἐν- — в- и ζύμη — дрожжи , закваска, то есть «в дрожжах») для обозначения «неорганизованных ферментов», секретируемых клетками, например, в желудок ( пепсин ) или кишечник ( трипсин , амилаза ).
Через два года после смерти Л. Пастера в 1897 году Э. Бухнер опубликовал работу «Спиртовое брожение без дрожжевых клеток», в которой экспериментально показал, что бесклеточный дрожжевой сок осуществляет спиртовое брожение так же, как и неразрушенные дрожжевые клетки. В 1907 году за эту работу он был удостоен Нобелевской премии. Впервые высокоочищенный кристаллический фермент ( уреаза ) был выделен в 1926 году Дж. Самнером . В течение последующих 10 лет было выделено ещё несколько ферментов, и белковая природа ферментов была окончательно доказана.
Каталитическая активность РНК впервые была обнаружена в 1980-е годы у пре-рРНК Томасом Чеком , изучавшим сплайсинг РНК у инфузории Tetrahymena thermophila . Рибозимом оказался участок молекулы пре-рРНК Tetrahymena, кодируемый интроном внехромосомного гена рДНК; этот участок осуществлял аутосплайсинг, то есть сам вырезал себя при созревании рРНК.
Существуют два основных пути повышения скорости химической реакции. Первый путь — повышение температуры, то есть ускорение теплового движения молекул, которое приводит к увеличению доли молекул, обладающих достаточной внутренней энергией для достижения переходного состояния. Как правило, повышение температуры на 10 °C вызывает ускорение химической реакции приблизительно в 2 раза (см. правило Вант-Гоффа ).

Второй путь ускорения химической реакции — добавление катализатора. Катализаторы ускоряют химические реакции, находя «обходные пути», позволяющие молекулам преодолевать активационный барьер на более низком энергетическом уровне. Катализатор (обозначим его буквой К ) на промежуточной стадии взаимодействует с реагентом А с образованием нового комплексного соединения КА , переходному состоянию которого соответствует значительно более низкая энергия активации по сравнению с переходным состоянием реагента А в некатализируемой реакции. Затем комплекс реагент-катализатор ( КА ) распадается на продукт П и свободный катализатор, который может опять соединиться с другой молекулой А и повторить весь цикл. Именно таким образом катализаторы снижают энергию активации химической реакции, в их присутствии гораздо более значительная доля молекул данной популяции вступает в реакцию в единицу времени. Ферменты, так же как и другие катализаторы, соединяются со своими субстратами в ходе каталитического цикла .
Ферменты присутствуют во всех живых клетках и способствуют превращению одних веществ в другие. Ферменты выступают в роли катализаторов практически во всех биохимических реакциях, протекающих в живых организмах. К 2013 году было описано более 5000 разных ферментов . Они играют важнейшую роль во всех процессах жизнедеятельности, направляя и регулируя обмен веществ организма.
Подобно всем катализаторам, ферменты ускоряют как прямую, так и обратную реакцию, понижая энергию активации процесса. Химическое равновесие при этом не смещается ни в прямую, ни в обратную сторону. Отличительной особенностью ферментов по сравнению с небелковыми катализаторами является их высокая — некоторых субстратов с белком может достигать 10 −10 моль/л и менее. Каждая молекула фермента способна выполнять от нескольких тысяч до нескольких миллионов «операций» в секунду.
Например, одна молекула фермента реннина , содержащегося в слизистой оболочке желудка телёнка, створаживает около 10 6 молекул казеиногена молока за 10 мин при температуре 37 °C.
При этом эффективность ферментов значительно выше эффективности небелковых катализаторов — ферменты ускоряют реакцию в миллионы и миллиарды раз, небелковые катализаторы — в сотни и тысячи раз. (см. также Каталитически совершенный фермент )
Обычно ферменты именуют по типу катализируемой реакции, добавляя суффикс -аза к названию субстрата ( например , лактаза — фермент, участвующий в превращении лактозы ). Таким образом, у различных ферментов, выполняющих одну функцию, будет одинаковое название или один и тот же фермент имеет два и более названий. Такие ферменты различают по другим свойствам, например, по оптимальному pH ( щелочная фосфатаза ) или локализации в клетке (мембранная АТФаза ). Многие ферменты имеют исторически сложившиеся тривиальные названия, не связанные с названиями их субстратов, например пепсин и трипсин . Из-за этих и других затруднений, а также вследствие всевозрастающего числа вновь открываемых ферментов было принято международное соглашение о создании систематической номенклатуры и классификации ферментов .
По типу катализируемых реакций ферменты подразделяются на 7 классов согласно иерархической классификации ферментов ( КФ , EC — Enzyme Comission code). Классификация была предложена Международным союзом биохимии и молекулярной биологии ( ). Каждый класс содержит подклассы, так что фермент описывается совокупностью четырёх чисел, разделённых точками. Например, пепсин имеет название ЕС 3.4.23.1. Первое число грубо описывает механизм реакции, катализируемой ферментом:
Второе число в наименовании фермента отражает подкласс, третье — подподкласс, а четвёртое — порядковый номер фермента в его подподклассе .
Будучи катализаторами , все ферменты ускоряют как прямую, так и обратную реакции, поэтому, например, лиазы способны катализировать и обратную реакцию — присоединение по двойным связям.

Простейшим описанием кинетики односубстратных ферментативных реакций является уравнение Михаэлиса — Ментен (см. рис.).
В 1972—1973 гг. была создана первая квантово-механическая модель ферментативного катализа (авторы М. В. Волькенштейн , Р. Р. Догонадзе, З. Д. Урушадзе и др.) . Предположим, концентрация фермента постоянна и необходимо измерить влияние изменения концентрации субстрата на начальную скорость ферментативной реакции. При очень низких концентрациях субстрата скорость реакции очень мала, но стабильно возрастает по мере постепенного повышения концентрации субстрата. Однако приращения скорости каталитической реакции становятся с каждым возрастанием концентрации субстрата всё меньше и меньше. Наконец, наступает момент, когда любое увеличение концентрации субстрата вызывает лишь бесконечно малое ускорение реакции: как бы ни увеличивалась концентрация субстрата, скорость реакции может лишь приближаться к плато, но никогда его не достигнет. На этом плато, называемом максимальной скоростью реакции ( V max ), фермент насыщен субстратом и не может функционировать быстрее. Данный эффект насыщения свойственен почти всем ферментам.
Величину V max можно определить из представленного графика путём аппроксимирования. Точное определение в данном случае невозможно, так как по мере повышения концентрации субстрата начальная скорость реакции лишь приближается к V max , но никогда её не достигает. Концентрация субстрата, при которой скорость реакции составляет половину максимальной (на графике обозначена как ½ V max ), представляет собой константу Михаэлиса — Ментен ( K M ). Её можно определить либо из графика, также путём аппроксимирования, либо путём алгебраических преобразований уравнения Михаэлиса — Ментен .
Активность ферментов определяется их трёх- и четырёхмерной структурой .
Как и все белки, ферменты синтезируются в виде линейной цепочки аминокислот , которая сворачивается определённым образом. Каждая последовательность аминокислот сворачивается особым образом, и получающаяся молекула ( белковая глобула ) обладает уникальными свойствами. Несколько белковых цепей могут объединяться в белковый комплекс . Третичная и четвертичная структуры белков разрушается при нагревании, изменении pH или воздействии некоторых химических веществ.
На сегодняшний момент описано несколько механизмов действия ферментов. В простой ферментативной реакции может участвовать только одна молекула субстрата С, связывающаяся с ферментом Ф с образованием продукта П:
Однако на самом деле во многих ферментативных реакциях метаболизма принимают участие и связываются с ферментом две, а иногда даже три молекулы разных субстратов. Такие реакции обычно включают перенос атома или функциональной группы от одного субстрата к другому. Такие реакции могут протекать по двум различным механизмам. В реакциях первого типа, называемых реакциями единичного замещения , два субстрата С 1 и С 2 связываются с ферментом Ф либо специфическим, либо случайным образом с образованием комплекса Ф С 1 С 2 , который затем распадается на продукты П 1 и П 2 :
Второй класс двухсубстратных реакций составляют реакции, протекающие по механизму двойного замещения (механизм типа «пинг-понг»):
В этих реакциях с каталитическим центром фермента в данный момент времени связан только один из двух субстратов. Присоединение первого субстрата сопровождается переносом его функциональной группы на молекулу фермента. Только после удаления продукта, образовавшегося из первого субстрата, второй субстрат может связаться с ферментом и принять функциональную группу .
Изучение механизма химической реакции , катализируемой ферментом, наряду с определением промежуточных и конечных продуктов на разных стадиях реакции подразумевает точное знание геометрии третичной структуры фермента, природы функциональных групп его молекулы , обеспечивающих специфичность действия и высокую каталитическую активность на данный субстрат , а также химической природы участка (участков) молекулы фермента, который обеспечивает высокую скорость каталитической реакции. Обычно молекулы субстрата, участвующие в ферментативных реакциях, по сравнению с молекулами ферментов имеют относительно небольшие размеры. Таким образом, при образовании фермент-субстратных комплексов в непосредственное химическое взаимодействие вступают лишь ограниченные фрагменты аминокислотной последовательности полипептидной цепи — «активный центр» — уникальная комбинация остатков аминокислот в молекуле фермента, обеспечивающая непосредственное взаимодействие с молекулой субстрата и прямое участие в акте катализа .
В активном центре условно выделяют :
Чтобы катализировать реакцию, фермент должен связаться с одним или несколькими субстратами. Белковая цепь фермента сворачивается таким образом, что на поверхности глобулы образуется щель, или впадина, где связываются субстраты. Эта область называется сайтом связывания субстрата. Обычно он совпадает с активным центром фермента или находится вблизи него. Некоторые ферменты содержат также сайты связывания кофакторов или ионов металлов.
Фермент, соединяясь с субстратом:
Обычно присоединение фермента к субстрату происходит за счёт ионных или водородных связей, редко — за счёт ковалентных. В конце реакции её продукт (или продукты) отделяются от фермента.
В результате фермент снижает энергию активации реакции. Это происходит потому, что в присутствии фермента реакция идёт по другому пути (фактически происходит другая реакция), например:
В отсутствие фермента:
В присутствии фермента:
где А, В — субстраты, АВ — продукт реакции, Ф — фермент.
Ферменты не могут самостоятельно обеспечивать энергией эндергонические реакции (для протекания которых требуется энергия). Поэтому ферменты, осуществляющие такие реакции, сопрягают их с экзергоническими реакциями, идущими с выделением большего количества энергии. Например, реакции синтеза биополимеров часто сопрягаются с реакцией гидролиза АТФ.
Для активных центров некоторых ферментов характерно явление кооперативности .
Ферменты обычно проявляют высокую специфичность по отношению к своим субстратам (субстратная специфичность). Это достигается частичной комплементарностью формы, распределения зарядов и гидрофобных областей на молекуле субстрата и в центре связывания субстрата на ферменте. Ферменты обычно демонстрируют также высокий уровень стереоспецифичности (образуют в качестве продукта только один из возможных стереоизомеров или используют в качестве субстрата только один стереоизомер), региоселективности (образуют или разрывают химическую связь только в одном из возможных положений субстрата) и хемоселективности (катализируют только одну химическую реакцию из нескольких возможных для данных условий). Несмотря на общий высокий уровень специфичности, степень субстратной и реакционной специфичности ферментов может быть различной. Например, эндопептидаза трипсин разрывает пептидную связь только после аргинина или лизина , если за ними не следует пролин, а пепсин гораздо менее специфичен и может разрывать пептидную связь, следующую за многими аминокислотами.


В 1890 г. Эмиль Фишер предположил, что специфичность ферментов определяется точным соответствием формы фермента и субстрата . Такое предположение называется моделью «ключ-замок». Фермент соединяется с субстратом с образованием короткоживущего фермент-субстратного комплекса. Однако, хотя эта модель объясняет высокую специфичность ферментов, она не объясняет явления стабилизации переходного состояния, которое наблюдается в эксперименте.
В 1958 г. Дениел Кошланд предложил модификацию модели «рука-перчатка» . Ферменты, в основном, — не жёсткие, а гибкие молекулы. Активный центр фермента может изменить конформацию после связывания субстрата. Боковые группы аминокислот активного центра принимают такое положение, которое позволяет ферменту выполнить свою каталитическую функцию. В некоторых случаях молекула субстрата также меняет конформацию после связывания в активном центре. В отличие от модели «ключ-замок», модель индуцированного соответствия объясняет не только специфичность ферментов, но и стабилизацию переходного состояния. Эта модель получила название «рука-перчатка».
Многие ферменты после синтеза белковой цепи претерпевают модификации, без которых фермент не проявляет свою активность в полной мере. Такие модификации называются посттрансляционными модификациями (процессингом). Один из самых распространённых типов модификации — присоединение химических групп к боковым остаткам полипептидной цепи. Например, присоединение остатка фосфорной кислоты называется фосфорилированием, оно катализируется ферментом киназой . Многие ферменты эукариот гликозилированы, то есть модифицированы олигомерами углеводной природы.
Ещё один распространённый тип посттрансляционных модификаций — расщепление полипептидной цепи. Например, ( протеаза , участвующая в пищеварении ), получается при выщеплении полипептидного участка из химотрипсиногена. Химотрипсиноген является неактивным предшественником химотрипсина и синтезируется в поджелудочной железе , а оттуда транспортируется в двенадцатиперстную кишку , где эта неактивная форма активируется трипсином и превращается в химотрипсин.
Некоторые ферменты выполняют каталитическую функцию сами по себе, безо всяких дополнительных компонентов. Однако есть ферменты, которым для осуществления катализа необходимы компоненты небелковой природы. Кофакторы могут быть как неорганическими молекулами (ионы металлов, железо-серные кластеры и др.), так и органическими (например, флавин или гем ). Органические кофакторы, прочно связанные с ферментом, называют также простетическими группами. Кофакторы органической природы, способные отделяться от фермента, называют коферментами.
Фермент, который требует наличия кофактора для проявления каталитической активности, но не связан с ним, называется апо-фермент. Апо-фермент в комплексе с кофактором носит название холо-фермента. Большинство кофакторов связано с ферментом нековалентными, но довольно прочными взаимодействиями. Есть и такие простетические группы, которые связаны с ферментом ковалентно, например, тиаминпирофосфат в пируватдегидрогеназе.

Активность ферментов зависит от условий в клетке или организме — давления, кислотности среды, температуры, концентрации растворённых солей (ионной силы раствора) и др.
Активность ферментов непостоянна во времени. Они чутко реагируют на ситуацию, в которой оказывается клетка, на факторы, воздействующие на неё как снаружи, так и изнутри. Главная цель такой чувствительности ферментов — отреагировать на изменение окружающей среды, приспособить клетку к новым условиям, дать должный ответ на гормональные и иные стимулы, а в некоторых ситуациях — получить шанс выжить .

Действие большинства ферментов можно подавить, или ингибировать , определёнными химическими реагентами. Механизм действия некоторых лекарственных препаратов состоит именно в том, что они ингибируют определённые ферменты в клетках с нарушенными функциями.
Существуют ингибиторы двух основных типов: необратимые и обратимые. Обратимые ингибиторы связываются с ферментом слабыми нековалентными связями и при определённых условиях легко отделяются . Необратимые ингибиторы связывают или разрушают функциональную группу молекулы фермента. Например, диизопропилфторфосфат (ДФФ, одно из первых отравляющих веществ нервно-паралитического действия) присоединяется к OH-группе остатка серина в активном центре ацетилхолинэстеразы, играющей важную роль в передаче нервных импульсов, и фермент перестаёт функционировать. ДФФ способен ингибировать целый класс ферментов, катализирующих гидролиз пептидов или эфирных связей, куда помимо ацетилхолинэстеразы входят трипсин , химотрипсин, эластаза, фосфоглюкомутаза и коконаза (фермент, выделяемый личинкой шелкопряда для гидролиза шёлковых нитей и освобождения из кокона). Характерной особенностью всех этих ферментов является наличие остатка серина в активном центре. Другой необратимый ингибитор, йодацетамид, может взаимодействовать с SH-группами остатков цистеина или с имидазольными группами остатков гистидина , содержащихся в активных центрах другого ряда ферментов.
Обратимые ингибиторы по своей природе бывают конкурентными, неконкурентными и бесконкурентными. Конкурентный ингибитор конкурирует с субстратом за связывание с активным центром, но в отличие от субстрата связанный с ферментом конкурентный ингибитор не подвергается ферментативному превращению. Отличительная особенность конкурентного ингибирования состоит в том, что его можно ослабить или вовсе устранить, просто повысив концентрацию субстрата. По своей трёхмерной структуре конкурентные ингибиторы обычно напоминают субстрат данного фермента. Благодаря такому сходству они «обманывают» фермент и связываются с ним. Классическим примером может служить ингибирование сукцинатдегидрогеназы анионом малоновой кислоты ( − OOC—CH 2 —COO − ), который напоминает сукцинат ( − OOC—CH 2 —CH 2 —COO − ) тем, что также содержит две близко расположенные карбоксильные группы, принимающие при pH = 7,0 ионизированную (депротонированную) форму, однако содержит 3, а не 4 атома углерода. Сукцинатдегидрогеназа не способна отщеплять водород от малоната, но малонат занимает активный центр фермента, не давая ему возможности взаимодействовать с нормальным субстратом. Повышение концентрации сукцината при фиксированной концентрации малоната снижает степень ингибирования фермента. Также конкурентным ингибитором сукцинатдегидрогеназы выступает оксалоацетат ( − OOC—CO—CH 2 —COO − ).
В случае неконкурентного ингибирования вещество присоединяется к ферменту не в активном центре, а совсем в другом месте, однако при этом конформация молекулы фермента изменяется таким образом, что происходит обратимая инактивация его каталитического центра. Неконкурентные ингибиторы связываются обратимо как со свободным ферментом, так и с фермент-субстратным комплексом, образуя неактивные комплексы фермент-ингибитор и фермент-субстрат-ингибитор. Наиболее важные неконкурентные ингибиторы представляют собой образующиеся в живых организмах промежуточные продукты метаболизма, способные обратимо связываться со специфическими участками на поверхности регуляторных ферментов. Примером может служить ингибирование L-треониндегидратазы L-изолейцином .
При бесконкурентном ингибировании ингибитор связывается только с фермент-субстратным комплексом, но не со свободным ферментом. Субстрат, связываясь с ферментом, изменяет его конформацию, что делает возможным связывание с ингибитором. Ингибитор, в свою очередь, так меняет конформацию фермента, что дальнейший катализ становится невозможным.
Метаболический путь — цепочка последовательных ферментативных реакций. В некоторых случаях молекулы конечного продукта метаболического пути связываются с продуцировавшим их ферментом и препятствуют дальнейшему образованию этого же самого продукта, таким образом конечный продукт тоже может являться ингибитором фермента. Такая ситуация является частным случаем бесконкурентного ингибирования. Как правило, в таких случаях речь идёт о блокировании самой первой реакции данного метаболического пути. Если конечного продукта слишком много, то он действует как ингибитор для самого первого фермента, а если после этого конечного продукта стало слишком мало, то первый фермент опять активируется (продукты метаболического пути в этом случае сами оказываются субстратами). Так ингибирование конечным продуктом создаёт возможность для осуществления отрицательной обратной связи — важного способа поддержания гомеостаза (относительного постоянства условий внутренней среды организма), а регуляция такого типа называется ингибированием по принципу обратной связи , или ретроингибированием . Классическим примером подобного ингибирования может служить бактериальная ферментная система, катализирующая превращение L-треонина в L-изолейцин под воздействием треониндегидратазы — процесс, включающий 5 ферментативных реакций. Треониндегидратаза ингибируется продуктом последней реакции — изолейцином, причём изолейцин связывается не с субстратным центром фермента, а с другим специфическим участком его молекулы, называемым регуляторным центром . Это взаимодействие не сопровождается образованием прочных ковалентных связей и поэтому легко обратимо. Ни один из промежуточных продуктов этой цепи реакций не ингибирует треониндегидратазу и ни один другой фермент цепи не ингибируется изолейцином, поэтому его можно причислить к высокоспецифическим ингибиторам треониндегидратазы .
Во многих ситуациях действие ферментов становится необходимым увеличить, то есть совершить активацию ферментов. Активаторы — разнообразные вещества органической и неорганической природы, которые повышают скорость ферментативных реакций.
Примеры активаторов органической природы: жёлчные кислоты (активируют поджелудочную липазу), энтерокиназа (активирует трипсиноген), глутатион, цистеин, витамин С (повышают активность оксидоредуктаз), некоторые тканевые ферменты (оксидоредуктазы, катепсины, аргиназа, растительная протеиназа и др.) в значительной степени активируются соединениями, содержащими свободные SH-группы (глутатион, цистеин).
Примеры активаторов неорганической природы: HCl активирует пепсиноген, ионы металлов (Na + , Cl − , K + , Mg 2+ , Mn 2+ , Zn 2+ ) активируют очень многие ферменты, поскольку:
Особенно часто активаторами выступают ионы двухвалентных и, реже, одновалентных металлов. Получены доказательства, что около четверти всех известных ферментов для проявления полной каталитической активности нуждаются в присутствии металлов, без которых вообще очень многие ферменты становятся неактивными. Так, при удалении цинка угольная ангидраза (карбоангидраза), катализирующая биосинтез и распад Н 2 СО 3 , практически теряет свою ферментативную активность; более того, цинк при этом не может быть заменён никаким другим металлом. Известны ферменты, действие которых активируется ионами нескольких металлов; в частности, енолаза активируется Mg 2+ , Mn 2+ , К + . В ряде случаев ионы металлов (Со 2+ , Mg 2+ , Zn 2+ , Fe 2+ ) выполняют функции простетических групп ферментов, или служат акцепторами и донаторами электронов, или выступают в качестве электрофилов либо нуклеофилов, сохраняя реактивные группы в необходимой ориентации. В других случаях они способствуют присоединению субстрата к активному центру и образованию фермент-субстратного комплекса. Например, ионы Mg 2+ через отрицательно заряженную фосфатную группу обеспечивают присоединение монофосфатных эфиров органических веществ к активному центру фосфатаз, катализирующих гидролиз этих соединений. Иногда металл соединяется с субстратом, образуя истинный субстрат, на который действует фермент. В частности, ионы Mg 2+ активируют креатинфосфокиназу благодаря образованию истинного субстрата — магниевой соли АТФ. Наконец, имеются экспериментальные доказательства прямого участия металлов (например, ионов Са 2+ в молекуле амилазы слюны) в формировании и стабилизации активного центра и всей трёхмерной структуры молекулы фермента. Следует отметить также, что металлы нередко выступают в роли аллостерических модуляторов (эффекторов). Взаимодействуя с аллостерическим центром, подобный эффектор способствует образованию наиболее выгодной пространственной конфигурации фермента и активного фермент-субстратного комплекса.
Анионы в физиологических концентрациях обычно неэффективны или оказывают небольшое активирующее влияние на ферменты. Исключение составляют пепсин, некоторые оксидоредуктазы, активируемые анионами, а также амилаза слюны, катализирующая гидролиз крахмала, активность которой повышается при действии ионов Cl − , и аденилатциклаза, которая активируется анионами галогенов .
Существуют представители класса регуляторных ферментов, у которых переход активной формы в неактивную происходит путём ковалентной модификации молекулы фермента. К этому классу относится, например, гликогенфосфорилаза из мышц и печени, катализирующая реакцию отщепления глюкозы от гликогена:
Гликогенфосфорилаза существует в двух формах: в виде фосфорилазы a (активная форма) и фосфорилазы b (относительно неактивная форма). Фосфорилаза a представляет собой димер, состоящий из двух идентичных субъединиц, в каждой из которых имеется один специфический остаток серина , фосфорилированный по гидроксильной группе. Эти остатки фосфосерина необходимы для максимальной активности фермента. Эти фосфатные группы серина можно удалить с помощью фермента, называемого фосфатазой фосфорилазы , с образованием фосфорилазы b , гораздо менее активно катализирующей распад гликогена. Таким образом, активная форма гликогенфосфорилазы превращается в относительно неактивную форму в результате расщепления двух ковалентных связей между остатками фосфорной кислоты и двумя специфическими остатками серина в молекуле фермента.
Фосфорилаза b может снова реактивироваться, то есть превратиться в активную фосфорилазу a . Эта реакция осуществляется с помощью другого фермента, называемого киназой фосфорилазы , который катализирует перенос фосфатных групп от АТФ к гидроксильным группам специфических остатков серина в фосфорилазе b .
Таким образом, распад гликогена в скелетных мышцах и печени регулируется путём изменения количественных соотношений активной и неактивной форм фермента. Переход из одной формы в другую сопровождается изменениями четвертичной структуры фермента, затрагивающими и его каталитический центр.
Хотя в большинстве известных случаев регуляция действия ферментов путём их ковалентной модификации осуществляется через фосфорилирование и дефосфорилирование специфических остатков серина, только что описанным на примере гликогенфосфорилазы, существуют и другие способы ковалентной модификации ферментов, например, метилирование определённых аминокислотных остатков, присоединение к ним аденилатных групп и иные пути.
Некоторые более сложные регуляторные ферменты модулируются ковалентными и нековалентными механизмами. Такие ферменты катализируют реакции, представляющие собой наиболее важные этапы метаболизма, поэтому они взаимодействуют со множеством регуляторных метаболитов, осуществляющих как аллостерическую, так и ковалентную модификацию этих ферментов. К подобным ферментам относится и только что рассмотренная гликогенфосфорилаза, поскольку помимо ковалентной модификации возможно также и нековалентное (аллостерическое) взаимодействие его с аденилатом, который является активирующим модулятором фосфорилазы b . Другой пример — глутаминсинтетаза E. coli , один из наиболее сложных регуляторных ферментов, взаимодействующий со многими аллостерическими регуляторами и регулирующийся также путём обратимой ковалентной модификации .
Множественные формы ферментов можно разделить на две категории:
Изоферменты — это ферменты, синтез которых кодируется разными генами, у них разная первичная структура и разные свойства, но они катализируют одну и ту же реакцию. Виды изоферментов:
Собственно множественные формы (истинные) — это ферменты, синтез которых кодируется одним и тем же аллелем одного и того же гена, у них одинаковая первичная структура и свойства, но после синтеза на рибосомах они подвергаются модификации и становятся разными, хотя и катализируют одну и ту же реакцию.
Изоферменты разные на генетическом уровне и отличаются от первичной последовательности, а истинные множественные формы становятся разными на посттрансляционном уровне.
В случае с прионами речь также может заходить о штаммах .
Подобно любому другому белку, ферменты изменяются с течением времени через мутации и расхождение последовательностей. Учитывая их центральную роль в метаболизме , эволюция ферментов играет решающую роль в адаптации организмов. Ключевой вопрос заключается в том, могут ли ферменты изменять свою ферментативную активность одновременно и каким образом это происходит. Общепризнано, что многие новые ферментативные активности развились в результате дупликации генов и мутации дубликатов, хотя эволюция может происходить и без дупликации. Одним из примеров фермента, изменившего свою активность, является предок (MAP) и креатинамидиногидролаза (креатиназа), которые явно гомологичны, но катализируют разные реакции (MAP удаляет аминотерминальный метионин в новых белках, в то время как креатиназа гидролизует креатин до саркозина и мочевины ). Кроме того, MAP зависит от ионов металлов, в то время как креатиназа нет, следовательно, это свойство также было потеряно с течением времени . Небольшие изменения ферментативной активности чрезвычайно распространены среди ферментов. В частности, специфичность связывания субстрата может легко и быстро изменяться при изменении отдельных аминокислот в сайтах связывания субстрата. Это часто наблюдается в основных классах ферментов, таких как киназы .
Искусственная (in vitro) эволюция в настоящее время широко используется для изменения активности или специфичности ферментов для их промышленного применения.
Ферменты широко используются в народном хозяйстве — пищевой, аграрной, текстильной промышленности, в фармакологии и медицине. Большинство лекарств влияют на течение ферментативных процессов в организме, запуская или приостанавливая те или иные реакции.
Связь между ферментами и наследственными болезнями обмена веществ была впервые установлена в 1910-е гг. Гэррод назвал заболевания, связанные с дефектами ферментов, «врождёнными ошибками метаболизма».
Если происходит мутация в гене , кодирующем определённый фермент, может измениться аминокислотная последовательность фермента. При этом в результате большинства мутаций его каталитическая активность снижается или полностью пропадает. Если организм получает два таких мутантных гена (по одному от каждого из родителей), в организме перестаёт идти химическая реакция, которую катализирует данный фермент. Например, появление альбиносов связано с прекращением выработки фермента тирозиназы, отвечающего за одну из стадий синтеза тёмного пигмента меланина . Фенилкетонурия связана с пониженной или отсутствующей активностью фермента фенилаланин-4-гидроксилазы в печени.
В настоящее время известны сотни наследственных заболеваний, связанные с дефектами ферментов. Разработаны методы лечения и профилактики многих из таких болезней.
Ферменты используются в диагностике заболеваний путём количественного определения самих ферментов в биологических жидкостях при патологии.
Существует большой градиент концентрации ферментов между внутриклеточными и внеклеточными частями тела. Поэтому любые, даже незначительные, повреждения клеток (иногда функциональные расстройства) приводят к выделению ферментов во внеклеточное пространство, откуда они поступают в кровь. Повышение уровня внутриклеточных ферментов в плазме крови прямо зависит от природы повреждающего воздействия, времени действия и степени повреждения биомембран клеток и субклеточных структур органов .
| Направление | Используемые ферменты | Применение |
|---|---|---|
| Биотопливная промышленность | Целлюлазы | Расщепление целлюлозы на сахара, которые могут быть ферментированы для получения целлюлозного этанола. |
| Лигниназы | Предварительная обработка биомассы для производства биотоплива . | |
| Биологическое моющее средство | Протеазы , амилазы , липазы | Удаление белковых, крахмальных, жирных или масляных пятен с белья и посуды . |
| Маннаназы | Удаление пищевых разводов с гуаровой камеди. | |
| Пивоваренная промышленность | Амилаза , глюканазы, протеазы | Расщепление полисахаридов и белков в солоде . |
| Бетаглюканазы | Улучшение фильтрационных характеристик сусла и пива . | |
| Амилоглюкозидаза и пуллуланазы | Производство низкокалорийного пива и регуляция ферментируемости . | |
| Ацетолактатдекарбоксилаза (ALDC) | Повышение эффективности ферментации за счёт снижения диацетила . | |
| Кулинарное использование | Папаин | Размягчение мяса для приготовления пищи . |
| Молочная промышленность | Реннин | Гидролиз белка в производстве сыров. |
| Липазы | Производство сыра камамбер и голубых сыров, таких как Рокфор. | |
| Пищевая промышленность | Амилазы | Производство сахара из крахмала , например, при приготовлении кукурузного сиропа с высоким содержанием фруктозы . |
| Протеазы | Снижение уровня белка в муке для приготовления печенья. | |
| Трипсин | Производство гипоаллергенного детского питания. | |
| Целлюлазы, пектиназы | Очистка фруктовых соков. | |
| Молекулярная биология | Нуклеазы , ДНК-лигаза и полимеразы | Использование рестриктаз и ПЦР для создания рекомбинантной ДНК. |
| Бумажная промышленность | Ксиланазы, гемицеллюлазы и лигнинпероксидазы | Удаление лигнина из крафт-бумаги . |
| Личная гигиена | Протеазы | Удаление белков с контактных линз для предотвращения инфекций. |
|
|
|
|---|---|
| Активность | |
| Регуляция | |
| Классификация | |
| Типы |
|